
A Guide to Metabolic Network Modeling for Plant Biology


Abstract
:1. Introduction
2. Recent Achievements in Modeling Plant Metabolism
2.1. Genome-Scale Modeling in Understanding Central Metabolism
2.2. New Insights into Carbon Flow in Distinct Photosynthesis Types
2.3. Elucidation of the Complex Regulation of Phenylalanine and Monolignol Pathway
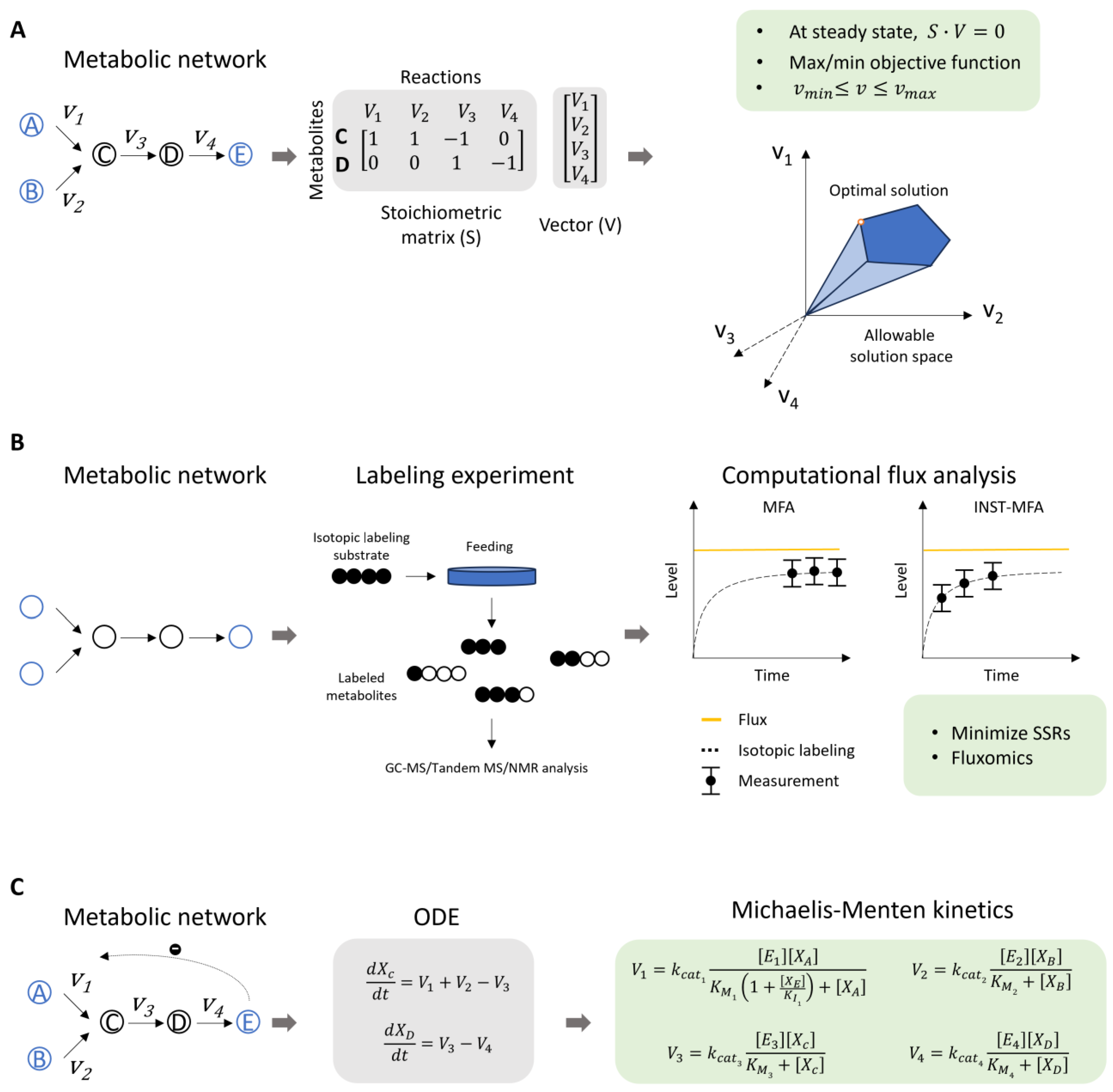
3. Strategies for Metabolic Model Reconstruction
3.1. Integration of Multi-Omics Data
3.2. A Proper Selection of Metabolic Model
4. Challenges in the Application of Metabolic Modeling in Plants
4.1. Incomplete Metabolic Pathways
4.2. High Degree of Subcellular Compartmentation
4.3. Cellular Heterogeneity
4.4. Incorporation of Multiscale Regulatory Processes
5. Advances in Machine Learning for Metabolic Modeling
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chandel, N.S. Basics of Metabolic Reactions. Cold Spring Harb. Perspect. Biol. 2021, 13, a040527. [Google Scholar] [CrossRef]
- Salam, U.; Ullah, S.; Tang, Z.H.; Elateeq, A.A.; Khan, Y.; Khan, J.; Khan, A.; Ali, S. Plant Metabolomics: An Overview of the Role of Primary and Secondary Metabolites against Different Environmental Stress Factors. Life 2023, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Obata, T. Metabolons in plant primary and secondary metabolism. Phytochem. Rev. 2019, 18, 1483–1507. [Google Scholar] [CrossRef]
- Wang, S.; Alseekh, S.; Fernie, A.R.; Luo, J. The Structure and Function of Major Plant Metabolite Modifications. Mol. Plant 2019, 12, 899–919. [Google Scholar] [CrossRef]
- Ort, D.R.; Merchant, S.S.; Alric, J.; Barkan, A.; Blankenship, R.E.; Bock, R.; Croce, R.; Hanson, M.R.; Hibberd, J.M.; Long, S.P.; et al. Redesigning photosynthesis to sustainably meet global food and bioenergy demand. Proc. Natl. Acad. Sci. USA 2015, 112, 8529–8536. [Google Scholar] [CrossRef]
- Benowitz, N.L. Pharmacology of nicotine: Addiction, smoking-induced disease, and therapeutics. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Naveen, J.; Baskaran, V. Antidiabetic plant-derived nutraceuticals: A critical review. Eur. J. Nutr. 2018, 57, 1275–1299. [Google Scholar] [CrossRef]
- Shih, M.L.; Morgan, J.A. Metabolic flux analysis of secondary metabolism in plants. Metab. Eng. Commun. 2020, 10, e00123. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.J.; Guo, L.; Morgan, J.; Schwender, J. Modeling Plant Metabolism: From Network Reconstruction to Mechanistic Models. Annu. Rev. Plant Biol. 2020, 71, 303–326. [Google Scholar] [CrossRef]
- Schwender, J. Metabolic flux analysis as a tool in metabolic engineering of plants. Curr. Opin. Biotechnol. 2008, 19, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Barros, J. Modeling lignin biosynthesis: A pathway to renewable chemicals. Trends Plant Sci. 2024, 29, 546–559. [Google Scholar] [CrossRef]
- Sweetlove, L.J.; Ratcliffe, R.G. Flux-balance modeling of plant metabolism. Front. Plant Sci. 2011, 2, 38. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, J.M. Kinetic modelling of plant metabolic pathways. J. Exp. Bot. 2012, 63, 2275–2292. [Google Scholar] [CrossRef]
- Koley, S.; Jyoti, P.; Lingwan, M.; Allen, D.K. Isotopically nonstationary metabolic flux analysis of plants: Recent progress and future opportunities. New Phytol. 2024, 242, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Gerlin, L.; Frainay, C.; Jourdan, F.; Baroukh, C.; Prigent, S. Chapter Eight—Plant genome-scale metabolic networks. In Advances in Botanical Research; Pétriacq, P., Bouchereau, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 237–270. [Google Scholar]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wondisford, F.E.; Song, C.; Zhang, T.; Su, X. Metabolic Flux Analysis-Linking Isotope Labeling and Metabolic Fluxes. Metabolites 2020, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Schroeder, W.L.; Saha, R. Kinetic modeling of metabolism: Present and future. Curr. Opin. Syst. Biol. 2021, 26, 72–78. [Google Scholar] [CrossRef]
- Fang, X.; Lloyd, C.J.; Palsson, B.O. Reconstructing organisms in silico: Genome-scale models and their emerging applications. Nat. Rev. Microbiol. 2020, 18, 731–743. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Tibocha-Bonilla, J.D.; Zuniga, C.; Godoy-Silva, R.D.; Zengler, K. Advances in metabolic modeling of oleaginous microalgae. Biotechnol. Biofuels 2018, 11, 241. [Google Scholar] [CrossRef]
- May, P.; Wienkoop, S.; Kempa, S.; Usadel, B.; Christian, N.; Rupprecht, J.; Weiss, J.; Recuenco-Munoz, L.; Ebenhoh, O.; Weckwerth, W.; et al. Metabolomics- and proteomics-assisted genome annotation and analysis of the draft metabolic network of Chlamydomonas reinhardtii. Genetics 2008, 179, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Boyle, N.R.; Morgan, J.A. Flux balance analysis of primary metabolism in Chlamydomonas reinhardtii. BMC Syst. Biol. 2009, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Dal’Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Nielsen, L.K. AlgaGEM—A genome-scale metabolic reconstruction of algae based on the Chlamydomonas reinhardtii genome. BMC Genom. 2011, 12 (Suppl. S4), S5. [Google Scholar]
- Cock, J.M.; Coelho, S.M. Algal models in plant biology. J. Exp. Bot. 2011, 62, 2425–2430. [Google Scholar] [CrossRef]
- Poolman, M.G.; Miguet, L.; Sweetlove, L.J.; Fell, D.A. A genome-scale metabolic model of Arabidopsis and some of its properties. Plant Physiol. 2009, 151, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Dal’Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Brumbley, S.M.; Nielsen, L.K. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010, 152, 579–589. [Google Scholar]
- Mintz-Oron, S.; Meir, S.; Malitsky, S.; Ruppin, E.; Aharoni, A.; Shlomi, T. Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc. Natl. Acad. Sci. USA 2012, 109, 339–344. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Williams, T.C.; Poolman, M.G.; Fell, D.A.; Ratcliffe, R.G.; Sweetlove, L.J. A method for accounting for maintenance costs in flux balance analysis improves the prediction of plant cell metabolic phenotypes under stress conditions. Plant J. 2013, 75, 1050–1061. [Google Scholar] [CrossRef]
- Dal’Molin, C.G.; Quek, L.E.; Saa, P.A.; Nielsen, L.K. A multi-tissue genome-scale metabolic modeling framework for the analysis of whole plant systems. Front. Plant Sci. 2015, 6, 4. [Google Scholar]
- Shaw, R.; Cheung, C.Y.M. A Dynamic Multi-Tissue Flux Balance Model Captures Carbon and Nitrogen Metabolism and Optimal Resource Partitioning During Arabidopsis Growth. Front. Plant Sci. 2018, 9, 884. [Google Scholar] [CrossRef] [PubMed]
- Pilalis, E.; Chatziioannou, A.; Thomasset, B.; Kolisis, F. An in silico compartmentalized metabolic model of Brassica napus enables the systemic study of regulatory aspects of plant central metabolism. Biotechnol. Bioeng. 2011, 108, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, M.; Zhang, Z.; Mohanty, B.; Kwon, J.Y.; Choi, H.Y.; Nam, H.J.; Kim, D.I.; Lee, D.Y. Elucidating rice cell metabolism under flooding and drought stresses using flux-based modeling and analysis. Plant Physiol. 2013, 162, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Poolman, M.G.; Kundu, S.; Shaw, R.; Fell, D.A. Responses to light intensity in a genome-scale model of rice metabolism. Plant Physiol. 2013, 162, 1060–1072. [Google Scholar] [CrossRef]
- Lakshmanan, M.; Lim, S.H.; Mohanty, B.; Kim, J.K.; Ha, S.H.; Lee, D.Y. Unraveling the Light-Specific Metabolic and Regulatory Signatures of Rice through Combined in Silico Modeling and Multiomics Analysis. Plant Physiol. 2015, 169, 3002–3020. [Google Scholar] [PubMed]
- Chatterjee, A.; Kundu, S. Revisiting the chlorophyll biosynthesis pathway using genome scale metabolic model of Oryza sativa japonica. Sci. Rep. 2015, 5, 14975. [Google Scholar] [CrossRef]
- Chatterjee, A.; Huma, B.; Shaw, R.; Kundu, S. Reconstruction of Oryza sativa indica Genome Scale Metabolic Model and Its Responses to Varying RuBisCO Activity, Light Intensity, and Enzymatic Cost Conditions. Front. Plant Sci. 2017, 8, 2060. [Google Scholar] [CrossRef] [PubMed]
- Dal’Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Brumbley, S.M.; Nielsen, L.K. C4GEM, a genome-scale metabolic model to study C4 plant metabolism. Plant Physiol. 2010, 154, 1871–1885. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Suthers, P.F.; Maranas, C.D. Zea mays iRS1563: A comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS ONE 2011, 6, e21784. [Google Scholar] [CrossRef] [PubMed]
- Dal’Molin, C.G.; Quek, L.E.; Saa, P.A.; Palfreyman, R.; Nielsen, L.K. From reconstruction to C4 metabolic engineering: A case study for overproduction of polyhydroxybutyrate in bioenergy grasses. Plant Sci. 2018, 273, 50–60. [Google Scholar] [CrossRef]
- Simons, M.; Saha, R.; Amiour, N.; Kumar, A.; Guillard, L.; Clement, G.; Miquel, M.; Li, Z.; Mouille, G.; Lea, P.J.; et al. Assessing the metabolic impact of nitrogen availability using a compartmentalized maize leaf genome-scale model. Plant Physiol. 2014, 166, 1659–1674. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.; Bradbury, L.M.; Frelin, O.; Zarecki, R.; Ruppin, E.; Hanson, A.D.; Henry, C.S. Improved evidence-based genome-scale metabolic models for maize leaf, embryo, and endosperm. Front. Plant Sci. 2015, 6, 142. [Google Scholar] [CrossRef]
- Bogart, E.; Myers, C.R. Multiscale Metabolic Modeling of C4 Plants: Connecting Nonlinear Genome-Scale Models to Leaf-Scale Metabolism in Developing Maize Leaves. PLoS ONE 2016, 11, e0151722. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, N.B.; Simons-Senftle, M.; Decouard, B.; Quillere, I.; Rigault, M.; Sajeevan, K.A.; Acharya, B.; Chowdhury, R.; Hirel, B.; Dellagi, A.; et al. A multi-organ maize metabolic model connects temperature stress with energy production and reducing power generation. iScience 2023, 26, 108400. [Google Scholar] [CrossRef] [PubMed]
- Dal’Molin, C.G.; Orellana, C.; Gebbie, L.; Steen, J.; Hodson, M.P.; Chrysanthopoulos, P.; Plan, M.R.; McQualter, R.; Palfreyman, R.W.; Nielsen, L.K. Metabolic Reconstruction of Setaria italica: A Systems Biology Approach for Integrating Tissue-Specific Omics and Pathway Analysis of Bioenergy Grasses. Front. Plant Sci. 2016, 7, 1138. [Google Scholar]
- Grafahrend-Belau, E.; Schreiber, F.; Koschutzki, D.; Junker, B.H. Flux balance analysis of barley seeds: A computational approach to study systemic properties of central metabolism. Plant Physiol. 2009, 149, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Lange, I.; Srividya, N.; Lange, B.M. Bioenergetics of Monoterpenoid Essential Oil Biosynthesis in Nonphotosynthetic Glandular Trichomes. Plant Physiol. 2017, 175, 681–695. [Google Scholar] [CrossRef]
- Moreira, T.B.; Shaw, R.; Luo, X.; Ganguly, O.; Kim, H.S.; Coelho, L.G.F.; Cheung, C.Y.M.; Rhys Williams, T.C. A Genome-Scale Metabolic Model of Soybean (Glycine max) Highlights Metabolic Fluxes in Seedlings. Plant Physiol. 2019, 180, 1912–1929. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Cheung, C.Y.; Poolman, M.G.; Hilbers, P.A.; van Riel, N.A. A genome-scale metabolic network reconstruction of tomato (Solanum lycopersicum L.) and its application to photorespiratory metabolism. Plant J. 2016, 85, 289–304. [Google Scholar] [CrossRef]
- Sarkar, D.; Maranas, C.D. SNPeffect: Identifying functional roles of SNPs using metabolic networks. Plant J. 2020, 103, 512–531. [Google Scholar] [CrossRef]
- Cunha, E.; Silva, M.; Chaves, I.; Demirci, H.; Lagoa, D.R.; Lima, D.; Rocha, M.; Rocha, I.; Dias, O. The first multi-tissue genome-scale metabolic model of a woody plant highlights suberin biosynthesis pathways in Quercus suber. PLoS Comput. Biol. 2023, 19, e1011499. [Google Scholar] [CrossRef] [PubMed]
- Botero, K.; Restrepo, S.; Pinzon, A. A genome-scale metabolic model of potato late blight suggests a photosynthesis suppression mechanism. BMC Genom. 2018, 19, 863. [Google Scholar] [CrossRef] [PubMed]
- Pfau, T.; Christian, N.; Masakapalli, S.K.; Sweetlove, L.J.; Poolman, M.G.; Ebenhoh, O. The intertwined metabolism during symbiotic nitrogen fixation elucidated by metabolic modelling. Sci. Rep. 2018, 8, 12504. [Google Scholar] [CrossRef]
- Luo, R.; Wei, H.; Ye, L.; Wang, K.; Chen, F.; Luo, L.; Liu, L.; Li, Y.; Crabbe, M.J.; Jin, L.; et al. Photosynthetic metabolism of C3 plants shows highly cooperative regulation under changing environments: A systems biological analysis. Proc. Natl. Acad. Sci. USA 2009, 106, 847–852. [Google Scholar] [CrossRef]
- Young, J.D.; Shastri, A.A.; Stephanopoulos, G.; Morgan, J.A. Mapping photoautotrophic metabolism with isotopically nonstationary 13C flux analysis. Metab. Eng. 2011, 13, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Jazmin, L.J.; Young, J.D.; Allen, D.K. Isotopically nonstationary 13C flux analysis of changes in Arabidopsis thaliana leaf metabolism due to high light acclimation. Proc. Natl. Acad. Sci. USA 2014, 111, 16967–16972. [Google Scholar] [CrossRef]
- Treves, H.; Kuken, A.; Arrivault, S.; Ishihara, H.; Hoppe, I.; Erban, A.; Hohne, M.; Moraes, T.A.; Kopka, J.; Szymanski, J.; et al. Carbon flux through photosynthesis and central carbon metabolism show distinct patterns between algae, C3 and C4 plants. Nat. Plants 2022, 8, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fu, X.; Sharkey, T.D.; Shachar-Hill, Y.; Walker, A.B.J. The metabolic origins of non-photorespiratory CO2 release during photosynthesis: A metabolic flux analysis. Plant Physiol. 2021, 186, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wieloch, T.; Kaste, J.A.M.; Shachar-Hill, Y.; Sharkey, T.D. Reimport of carbon from cytosolic and vacuolar sugar pools into the Calvin-Benson cycle explains photosynthesis labeling anomalies. Proc. Natl. Acad. Sci. USA 2022, 119, e2121531119. [Google Scholar] [CrossRef]
- Arrivault, S.; Obata, T.; Szecowka, M.; Mengin, V.; Guenther, M.; Hoehne, M.; Fernie, A.R.; Stitt, M. Metabolite pools and carbon flow during C4 photosynthesis in maize: 13CO2 labeling kinetics and cell type fractionation. J. Exp. Bot. 2017, 68, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brautigam, A.; Weber, A.P.; Zhu, X.G. Three distinct biochemical subtypes of C4 photosynthesis? A modelling analysis. J. Exp. Bot. 2014, 65, 3567–3578. [Google Scholar] [PubMed]
- Yin, X.; Struik, P.C. The energy budget in C4 photosynthesis: Insights from a cell-type-specific electron transport model. New Phytol. 2018, 218, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Struik, P.C. Exploiting differences in the energy budget among C4 subtypes to improve crop productivity. New Phytol. 2021, 229, 2400–2409. [Google Scholar] [CrossRef]
- Wang, Y.; Chan, K.X.; Long, S.P. Towards a dynamic photosynthesis model to guide yield improvement in C4 crops. Plant J. 2021, 107, 343–359. [Google Scholar] [CrossRef]
- Tay, I.Y.Y.; Odang, K.B.; Cheung, C.Y.M. Metabolic Modeling of the C3-CAM Continuum Revealed the Establishment of a Starch/Sugar-Malate Cycle in CAM Evolution. Front. Plant Sci. 2020, 11, 573197. [Google Scholar] [CrossRef] [PubMed]
- Shameer, S.; Baghalian, K.; Cheung, C.Y.M.; Ratcliffe, R.G.; Sweetlove, L.J. Computational analysis of the productivity potential of CAM. Nat. Plants 2018, 4, 165–171. [Google Scholar] [CrossRef]
- Topfer, N.; Braam, T.; Shameer, S.; Ratcliffe, R.G.; Sweetlove, L.J. Alternative Crassulacean Acid Metabolism Modes Provide Environment-Specific Water-Saving Benefits in a Leaf Metabolic Model. Plant Cell 2020, 32, 3689–3705. [Google Scholar] [CrossRef] [PubMed]
- Beilsmith, K.; Henry, C.S.; Seaver, S.M.D. Genome-scale modeling of the primary-specialized metabolism interface. Curr. Opin. Plant Biol. 2022, 68, 102244. [Google Scholar] [CrossRef]
- Allen, D.K. Quantifying plant phenotypes with isotopic labeling & metabolic flux analysis. Curr. Opin. Biotechnol. 2016, 37, 45–52. [Google Scholar]
- Lynch, J.H.; Orlova, I.; Zhao, C.; Guo, L.; Jaini, R.; Maeda, H.; Akhtar, T.; Cruz-Lebron, J.; Rhodes, D.; Morgan, J.; et al. Multifaceted plant responses to circumvent Phe hyperaccumulation by downregulation of flux through the shikimate pathway and by vacuolar Phe sequestration. Plant J. 2017, 92, 939–950. [Google Scholar] [CrossRef]
- Yoo, H.; Shrivastava, S.; Lynch, J.H.; Huang, X.Q.; Widhalm, J.R.; Guo, L.; Carter, B.C.; Qian, Y.; Maeda, H.A.; Ogas, J.P.; et al. Overexpression of arogenate dehydratase reveals an upstream point of metabolic control in phenylalanine biosynthesis. Plant J. 2021, 108, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Widhalm, J.R.; Gutensohn, M.; Yoo, H.; Adebesin, F.; Qian, Y.; Guo, L.; Jaini, R.; Lynch, J.H.; McCoy, R.M.; Shreve, J.T.; et al. Identification of a plastidial phenylalanine exporter that influences flux distribution through the phenylalanine biosynthetic network. Nat. Commun. 2015, 6, 8142. [Google Scholar] [CrossRef]
- Guo, L.; Wang, P.; Jaini, R.; Dudareva, N.; Chapple, C.; Morgan, J.A. Dynamic modeling of subcellular phenylpropanoid metabolism in Arabidopsis lignifying cells. Metab. Eng. 2018, 49, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Wang, X.; Docampo-Palacios, M.; Sanders, B.C.; Engle, N.L.; Tschaplinski, T.J.; Hendry, J.I.; Maranas, C.D.; Chen, F.; Dixon, R.A. Developmental changes in lignin composition are driven by both monolignol supply and laccase specificity. Sci. Adv. 2022, 8, eabm8145. [Google Scholar] [CrossRef]
- Faraji, M.; Voit, E.O. Improving Bioenergy Crops through Dynamic Metabolic Modeling. Processes 2017, 5, 61. [Google Scholar] [CrossRef]
- Faraji, M.; Fonseca, L.L.; Escamilla-Trevino, L.; Barros-Rios, J.; Engle, N.; Yang, Z.K.; Tschaplinski, T.J.; Dixon, R.A.; Voit, E.O. Mathematical models of lignin biosynthesis. Biotechnol. Biofuels 2018, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Faraji, M.; Fonseca, L.L.; Escamilla-Trevino, L.; Dixon, R.A.; Voit, E.O. Computational inference of the structure and regulation of the lignin pathway in Panicum virgatum. Biotechnol. Biofuels 2015, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chen, F.; Gallego-Giraldo, L.; Dixon, R.A.; Voit, E.O. Integrative analysis of transgenic alfalfa (Medicago sativa L.) suggests new metabolic control mechanisms for monolignol biosynthesis. PLoS Comput. Biol. 2011, 7, e1002047. [Google Scholar] [CrossRef]
- Wang, J.P.; Matthews, M.L.; Naik, P.P.; Williams, C.M.; Ducoste, J.J.; Sederoff, R.R.; Chiang, V.L. Flux modeling for monolignol biosynthesis. Curr. Opin. Biotechnol. 2019, 56, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Matthews, M.L.; Williams, C.M.; Shi, R.; Yang, C.; Tunlaya-Anukit, S.; Chen, H.-C.; Li, Q.; Liu, J.; Lin, C.-Y.; et al. Improving wood properties for wood utilization through multi-omics integration in lignin biosynthesis. Nat. Commun. 2018, 9, 1579. [Google Scholar]
- Wang, J.P.; Naik, P.P.; Chen, H.C.; Shi, R.; Lin, C.Y.; Liu, J.; Shuford, C.M.; Li, Q.; Sun, Y.H.; Tunlaya-Anukit, S.; et al. Complete proteomic-based enzyme reaction and inhibition kinetics reveal how monolignol biosynthetic enzyme families affect metabolic flux and lignin in Populus trichocarpa. Plant Cell 2014, 26, 894–914. [Google Scholar] [CrossRef]
- Volkova, S.; Matos, M.R.A.; Mattanovich, M.; Marin de Mas, I. Metabolic Modelling as a Framework for Metabolomics Data Integration and Analysis. Metabolites 2020, 10, 303. [Google Scholar] [CrossRef]
- Matthews, M.L.; Marshall-Colon, A. Multiscale plant modeling: From genome to phenome and beyond. Emerg. Top. Life Sci. 2021, 5, 231–237. [Google Scholar]
- Fondi, M.; Lio, P. Multi-omics and metabolic modelling pipelines: Challenges and tools for systems microbiology. Microbiol. Res. 2015, 171, 52–64. [Google Scholar] [CrossRef]
- Depuydt, T.; Vandepoele, K. Multi-omics network-based functional annotation of unknown Arabidopsis genes. Plant J. 2021, 108, 1193–1212. [Google Scholar] [CrossRef]
- Chen, C.; Liao, C.; Liu, Y.Y. Teasing out missing reactions in genome-scale metabolic networks through hypergraph learning. Nat. Commun. 2023, 14, 2375. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Shaked, I.; Oberhardt, M.A.; Atias, N.; Sharan, R.; Ruppin, E. Metabolic Network Prediction of Drug Side Effects. Cell Syst. 2016, 2, 209–213. [Google Scholar] [CrossRef]
- Fritzemeier, C.J.; Hartleb, D.; Szappanos, B.; Papp, B.; Lercher, M.J. Erroneous energy-generating cycles in published genome scale metabolic networks: Identification and removal. PLoS Comput. Biol. 2017, 13, e1005494. [Google Scholar] [CrossRef]
- Dal’molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Nielsen, L.K. Plant genome-scale modeling and implementation. Methods Mol. Biol. 2014, 1090, 317–332. [Google Scholar] [PubMed]
- Baghalian, K.; Hajirezaei, M.R.; Schreiber, F. Plant metabolic modeling: Achieving new insight into metabolism and metabolic engineering. Plant Cell 2014, 26, 3847–3866. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Segre, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J., 3rd. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Grafahrend-Belau, E.; Junker, A.; Eschenroder, A.; Muller, J.; Schreiber, F.; Junker, B.H. Multiscale metabolic modeling: Dynamic flux balance analysis on a whole-plant scale. Plant Physiol. 2013, 163, 637–647. [Google Scholar] [CrossRef]
- Zamboni, N.; Fendt, S.-M.; Rühl, M.; Sauer, U. 13C-based metabolic flux analysis. Nat. Protoc. 2009, 4, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.B.; Ahn, W.S.; Antoniewicz, M.R. Rational design of 13C-labeling experiments for metabolic flux analysis in mammalian cells. BMC Syst. Biol. 2012, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R. 13C metabolic flux analysis: Optimal design of isotopic labeling experiments. Curr. Opin. Biotechnol. 2013, 24, 1116–1121. [Google Scholar] [CrossRef]
- Radenkovic, S.; Vuckovic, I.; Lanza, I.R. Metabolic Flux Analysis: Moving beyond Static Metabolomics. Trends Biochem. Sci. 2020, 45, 545–546. [Google Scholar] [CrossRef] [PubMed]
- Wahrheit, J.; Nicolae, A.; Heinzle, E. Eukaryotic metabolism: Measuring compartment fluxes. Biotechnol. J. 2011, 6, 1071–1085. [Google Scholar] [CrossRef]
- Cheah, Y.E.; Young, J.D. Isotopically nonstationary metabolic flux analysis (INST-MFA): Putting theory into practice. Curr. Opin. Biotechnol. 2018, 54, 80–87. [Google Scholar] [CrossRef]
- Murabito, E.; Verma, M.; Bekker, M.; Bellomo, D.; Westerhoff, H.V.; Teusink, B.; Steuer, R. Monte-Carlo modeling of the central carbon metabolism of Lactococcus lactis: Insights into metabolic regulation. PLoS ONE 2014, 9, e106453. [Google Scholar] [CrossRef]
- Suthers, P.F.; Foster, C.J.; Sarkar, D.; Wang, L.; Maranas, C.D. Recent advances in constraint and machine learning-based metabolic modeling by leveraging stoichiometric balances, thermodynamic feasibility and kinetic law formalisms. Metab. Eng. 2021, 63, 13–33. [Google Scholar] [CrossRef]
- Shameer, S.; Wang, Y.; Bota, P.; Ratcliffe, R.G.; Long, S.P.; Sweetlove, L.J. A hybrid kinetic and constraint-based model of leaf metabolism allows predictions of metabolic fluxes in different environments. Plant J. 2022, 109, 295–313. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.K.; Libourel, I.G.; Shachar-Hill, Y. Metabolic flux analysis in plants: Coping with complexity. Plant Cell Environ. 2009, 32, 1241–1257. [Google Scholar] [CrossRef]
- Schlapfer, P.; Zhang, P.; Wang, C.; Kim, T.; Banf, M.; Chae, L.; Dreher, K.; Chavali, A.K.; Nilo-Poyanco, R.; Bernard, T.; et al. Genome-Wide Prediction of Metabolic Enzymes, Pathways, and Gene Clusters in Plants. Plant Physiol. 2017, 173, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yang, Y.; Gong, D.; Chen, X.; Jin, K.; Jiang, H.; Yu, W.; Li, J.; Zhang, J.; Pan, W. GFAP: Ultrafast and accurate gene functional annotation software for plants. Plant Physiol. 2023, 193, 1745–1748. [Google Scholar] [CrossRef]
- Moore, B.M.; Wang, P.; Fan, P.; Leong, B.; Schenck, C.A.; Lloyd, J.P.; Lehti-Shiu, M.D.; Last, R.L.; Pichersky, E.; Shiu, S.H. Robust predictions of specialized metabolism genes through machine learning. Proc. Natl. Acad. Sci. USA 2019, 116, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, L.J.; Fernie, A.R. The spatial organization of metabolism within the plant cell. Annu. Rev. Plant Biol. 2013, 64, 723–746. [Google Scholar] [CrossRef]
- Zecchin, A.; Stapor, P.C.; Goveia, J.; Carmeliet, P. Metabolic pathway compartmentalization: An underappreciated opportunity? Curr. Opin. Biotechnol. 2015, 34, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Furtauer, L.; Kustner, L.; Weckwerth, W.; Heyer, A.G.; Nagele, T. Resolving subcellular plant metabolism. Plant J. 2019, 100, 438–455. [Google Scholar] [CrossRef]
- Qin, S.; Zhang, Y.; Tian, Y.; Xu, F.; Zhang, P. Subcellular metabolomics: Isolation, measurement, and applications. J. Pharm. Biomed. Anal. 2022, 210, 114557. [Google Scholar] [CrossRef] [PubMed]
- Pandian, K.; Matsui, M.; Hankemeier, T.; Ali, A.; Okubo-Kurihara, E. Advances in single-cell metabolomics to unravel cellular heterogeneity in plant biology. Plant Physiol. 2023, 193, 949–965. [Google Scholar] [CrossRef]
- Yu, X.; Liu, Z.; Sun, X. Single-cell and spatial multi-omics in the plant sciences: Technical advances, applications, and perspectives. Plant Commun. 2023, 4, 100508. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.; Tian, X.; Xu, J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol. Plant 2021, 14, 115–126. [Google Scholar] [CrossRef]
- Dolatmoradi, M.; Samarah, L.Z.; Vertes, A. Single-Cell Metabolomics by Mass Spectrometry: Opportunities and Challenges. Anal. Sens. 2022, 2, e202100032. [Google Scholar] [CrossRef]
- Rosado-Souza, L.; Yokoyama, R.; Sonnewald, U.; Fernie, A.R. Understanding source-sink interactions: Progress in model plants and translational research to crops. Mol. Plant 2023, 16, 96–121. [Google Scholar] [CrossRef]
- Yu, S.M.; Lo, S.F.; Ho, T.D. Source-Sink Communication: Regulated by Hormone, Nutrient, and Stress Cross-Signaling. Trends Plant Sci. 2015, 20, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Maiti, R.; Shaw, R.; Cheung, C.Y.M.; Kundu, S. Metabolic modelling revealed source-sink interactions between four segments of Setaria viridis leaves. J. Biosci. 2023, 48, 26. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef]
- Kundu, P.; Beura, S.; Mondal, S.; Das, A.K.; Ghosh, A. Machine learning for the advancement of genome-scale metabolic modeling. Biotechnol. Adv. 2024, 74, 108400. [Google Scholar] [CrossRef]
- Peng, B.; Guan, K.; Tang, J.; Ainsworth, E.A.; Asseng, S.; Bernacchi, C.J.; Cooper, M.; Delucia, E.H.; Elliott, J.W.; Ewert, F.; et al. Towards a multiscale crop modelling framework for climate change adaptation assessment. Nat. Plants 2020, 6, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.; Cooper, M.; Tardieu, F.; Welch, S.; Walsh, B.; van Eeuwijk, F.; Chapman, S.; Podlich, D. Models for navigating biological complexity in breeding improved crop plants. Trends Plant Sci. 2006, 11, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, K.; Yang, F.; Pan, S.; Liu, Z.; Zhang, Q.; Zhang, Q. Prediction of maize cultivar yield based on machine learning algorithms for precise promotion and planting. Agric. For. Meteorol. 2024, 355, 110123. [Google Scholar] [CrossRef]
- Heinemann, A.B.; Costa-Neto, G.; da Matta, D.H.; Fernandes, I.K.; Stone, L.F. Harnessing crop models and machine learning for a spatial-temporal characterization of irrigated rice breeding environments in Brazil. Field Crops Res. 2024, 315, 109452. [Google Scholar] [CrossRef]
- Jaini, R. A Computational and Experimental Investigation of Lignin Metabolism in Arabidopsis thaliana. Ph.D. Thesis, Purdue University, West Lafayette, IN, USA, 2017. [Google Scholar]
- Xu, C.; Jackson, S.A. Machine learning and complex biological data. Genome Biol. 2019, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Geirhos, R.; Jacobsen, J.-H.; Michaelis, C.; Zemel, R.; Brendel, W.; Bethge, M.; Wichmann, F.A. Shortcut learning in deep neural networks. Nat. Mach. Intell. 2020, 2, 665–673. [Google Scholar] [CrossRef]
- Sampaio, M.; Rocha, M.; Dias, O. Exploring synergies between plant metabolic modelling and machine learning. Comput. Struct. Biotechnol. J. 2022, 20, 1885–1900. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Blatke, M.A.; Szymanski, J.J.; Topfer, N. Advances in flux balance analysis by integrating machine learning and mechanism-based models. Comput. Struct. Biotechnol. J. 2021, 19, 4626–4640. [Google Scholar] [CrossRef]
- Durand, M.; Besseau, S.; Papon, N.; Courdavault, V. Unlocking plant bioactive pathways: Omics data harnessing and machine learning assisting. Curr. Opin. Biotechnol. 2024, 87, 103135. [Google Scholar] [CrossRef] [PubMed]
- Duge de Bernonville, T.; Papon, N.; Clastre, M.; O’Connor, S.E.; Courdavault, V. Identifying Missing Biosynthesis Enzymes of Plant Natural Products. Trends Pharmacol. Sci. 2020, 41, 142–146. [Google Scholar] [CrossRef]
- Srinivasan, P.; Smolke, C.D. Engineering cellular metabolite transport for biosynthesis of computationally predicted tropane alkaloid derivatives in yeast. Proc. Natl. Acad. Sci. USA 2021, 118, e2104460118. [Google Scholar] [CrossRef] [PubMed]
- Duge de Bernonville, T.; Amor Stander, E.; Duge de Bernonville, G.; Besseau, S.; Courdavault, V. Predicting Monoterpene Indole Alkaloid-Related Genes from Expression Data with Artificial Neural Networks. Methods Mol. Biol. 2022, 2505, 131–140. [Google Scholar]
- Vavricka, C.J.; Takahashi, S.; Watanabe, N.; Takenaka, M.; Matsuda, M.; Yoshida, T.; Suzuki, R.; Kiyota, H.; Li, J.; Minami, H.; et al. Machine learning discovery of missing links that mediate alternative branches to plant alkaloids. Nat. Commun. 2022, 13, 1405. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.E.; Marti, J.M.; Radivojevic, T.; Jonnalagadda, S.V.R.; Gentz, R.; Hillson, N.J.; Peisert, S.; Kim, J.; Simmons, B.A.; Petzold, C.J.; et al. Machine learning for metabolic engineering: A review. Metab. Eng. 2021, 63, 34–60. [Google Scholar] [CrossRef]
- Hawkins, C.; Ginzburg, D.; Zhao, K.; Dwyer, W.; Xue, B.; Xu, A.; Rice, S.; Cole, B.; Paley, S.; Karp, P.; et al. Plant Metabolic Network 15: A resource of genome-wide metabolism databases for 126 plants and algae. J. Integr. Plant Biol. 2021, 63, 1888–1905. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Hu, X.; Xu, Y.; Liu, M.; Liu, H.; Li, D.; Hu, L.; Wei, G.; Chen, W. PMhub 1.0: A comprehensive plant metabolome database. Nucleic Acids Res. 2024, 52, D1579–D1587. [Google Scholar]
- Costello, Z.; Martin, H.G. A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. NPJ Syst. Biol. Appl. 2018, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Bordbar, A.; Yurkovich, J.T.; Paglia, G.; Rolfsson, O.; Sigurjonsson, O.E.; Palsson, B.O. Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci. Rep. 2017, 7, 46249. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Blomberg, P.; Castillo, S.; Rousu, J. Principal metabolic flux mode analysis. Bioinformatics 2018, 34, 2409–2417. [Google Scholar] [CrossRef]
- Magnusdottir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jager, C.; Baginska, J.; Wilmes, P.; et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2017, 35, 81–89. [Google Scholar] [CrossRef]
- DiMucci, D.; Kon, M.; Segre, D. Machine Learning Reveals Missing Edges and Putative Interaction Mechanisms in Microbial Ecosystem Networks. mSystems 2018, 3, e00181-18. [Google Scholar] [CrossRef]
- Matthews, M.L.; Wang, J.P.; Sederoff, R.; Chiang, V.L.; Williams, C.M. Modeling cross-regulatory influences on monolignol transcripts and proteins under single and combinatorial gene knockdowns in Populus trichocarpa. PLoS Comput. Biol. 2020, 16, e1007197. [Google Scholar] [CrossRef]
- Matthews, M.L.; Wang, J.P.; Sederoff, R.; Chiang, V.L.; Williams, C.M. A multiscale model of lignin biosynthesis for predicting bioenergy traits in Populus trichocarpa. Comput. Struct. Biotechnol. J. 2021, 19, 168–182. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Plant Species | Genes | Metabolites | Reactions | Model Properties | Refs |
|---|---|---|---|---|---|
| Arabidopsis thaliana | - | 1253 | 1406 | The first GSM model in plants | [26] |
| 1419 | 1748 | 1567 | AraGEM for primary metabolism with cellular compartmentalized | [27,30] | |
| - | 1078 | 1363 | A compartmentalized and tissue-specific model for both primary and secondary metabolism | [28] | |
| 2857 | 2739 | 2769 | An improved model to predict metabolic phenotypes under stress conditions | [29] | |
| 4262 | 2864 | 2801 | An improved model based on available evidence | [42] | |
| - | 10,664 | 11,320 | A dynamic model to investigate the carbon and nitrogen metabolism during plant growth | [31] | |
| Oryza sativa (rice) | 248 | 371 | 326 | The first GSM model in rice for metabolism under flooding and drought | [33] |
| - | 1484 | 1736 | A model representing rice leaf in responses to light intensity | [34] | |
| 2164 | 1999 | 2283 | A model focus on light-specific metabolism and light-mediated regulation | [35] | |
| - | 1544 | 1721 | A leaf model focus on chlorophyll synthesis | [36] | |
| 3602 | 1330 | 1136 | A model of O.s. indica. | [37] | |
| Zea mays (maize) | 11,623 | 1755 | 1588 | C4GEM for C4 plant metabolism | [38] |
| 1563 | 1825 | 1985 | A comprehensive and compartmentalized model for both primary and specialized metabolism under different physiological conditions | [39] | |
| 5824 | 9153 | 8525 | A model for the maize leaf on C4 carbon fixation and nitrogen assimilation with the interactions between the bundle sheath and mesophyll cells | [41] | |
| 5540 | 2634 | 2629 | Organ- and tissue-specific models for maize leaf, embryo, and endosperm | [42] | |
| 5204 | 2725 | 2720 | A mesophyll-bundle sheath model for flux prediction in the developing leaf | [43] | |
| - | 22,265 | 22,232 | The largest maize multi-organ model to identify metabolic regulation under cold and heat stress | [44] | |
| Saccharum officinarum (sugarcane) | 3881 | 1755 | 1558 | C4GEM for C4 plant metabolism | [38] |
| Sorghum bicolor (sorghum) | 3557 | 1755 | 1588 | C4GEM for C4 plant metabolism | [38] |
| Setaria italica (foxtail millet) | 1860 | 1690 | 1515 | A model based on C4GEM for the metabolism of S. italica | [45] |
| Hordeum vulgare (barley) | - | 234 | 257 | A model of primary metabolism in the developing endosperm of barley | [46] |
| Glycine max (soybean) | 6127 | 2814 | 3001 | A cotyledons and hypocotyl/root axis model for metabolic fluxes in soybean seedling | [48] |
| Brassica napus (Rapeseed) | - | 262 | 313 | A multi-compartmental model for seed metabolism | [32] |
| Solanum lycopersicum L. (tomato) | 3410 | 1998 | 2143 | A tomato leaf model to describe metabolic changes under heterotrophic and phototrophic conditions | [49] |
| Solanum tuberosum (potato) | 2751 | 1938 | 2072 | A leaf model to simulate the metabolic response of late blight | [52] |
| Medicago truncatula | 3403 | 2780 | 2909 | A multi-tissue model to investigate the metabolism during symbiotic nitrogen fixation | [53] |
| Mentha × piperita (peppermint) | 757 | 466 | 624 | A model of specialized metabolism in glandular trichomes | [47] |
| Populus trichocarpa | 7188 | 2502 | 3282 | A metabolic model for prediction of SNP effect on carbon and energy partition | [50] |
| Quercus suber (cork oak) | 7871 | 6481 | 6231 | The first multi-tissue GSM model in woody plants | [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, X.; Liu, W. A Guide to Metabolic Network Modeling for Plant Biology. Plants 2025, 14, 484. https://doi.org/10.3390/plants14030484
Rao X, Liu W. A Guide to Metabolic Network Modeling for Plant Biology. Plants. 2025; 14(3):484. https://doi.org/10.3390/plants14030484
Chicago/Turabian StyleRao, Xiaolan, and Wei Liu. 2025. "A Guide to Metabolic Network Modeling for Plant Biology" Plants 14, no. 3: 484. https://doi.org/10.3390/plants14030484
APA StyleRao, X., & Liu, W. (2025). A Guide to Metabolic Network Modeling for Plant Biology. Plants, 14(3), 484. https://doi.org/10.3390/plants14030484