
Genome-Wide Identification of ATL Gene Family in Wheat and Their Expression Analysis in Response to Salt Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
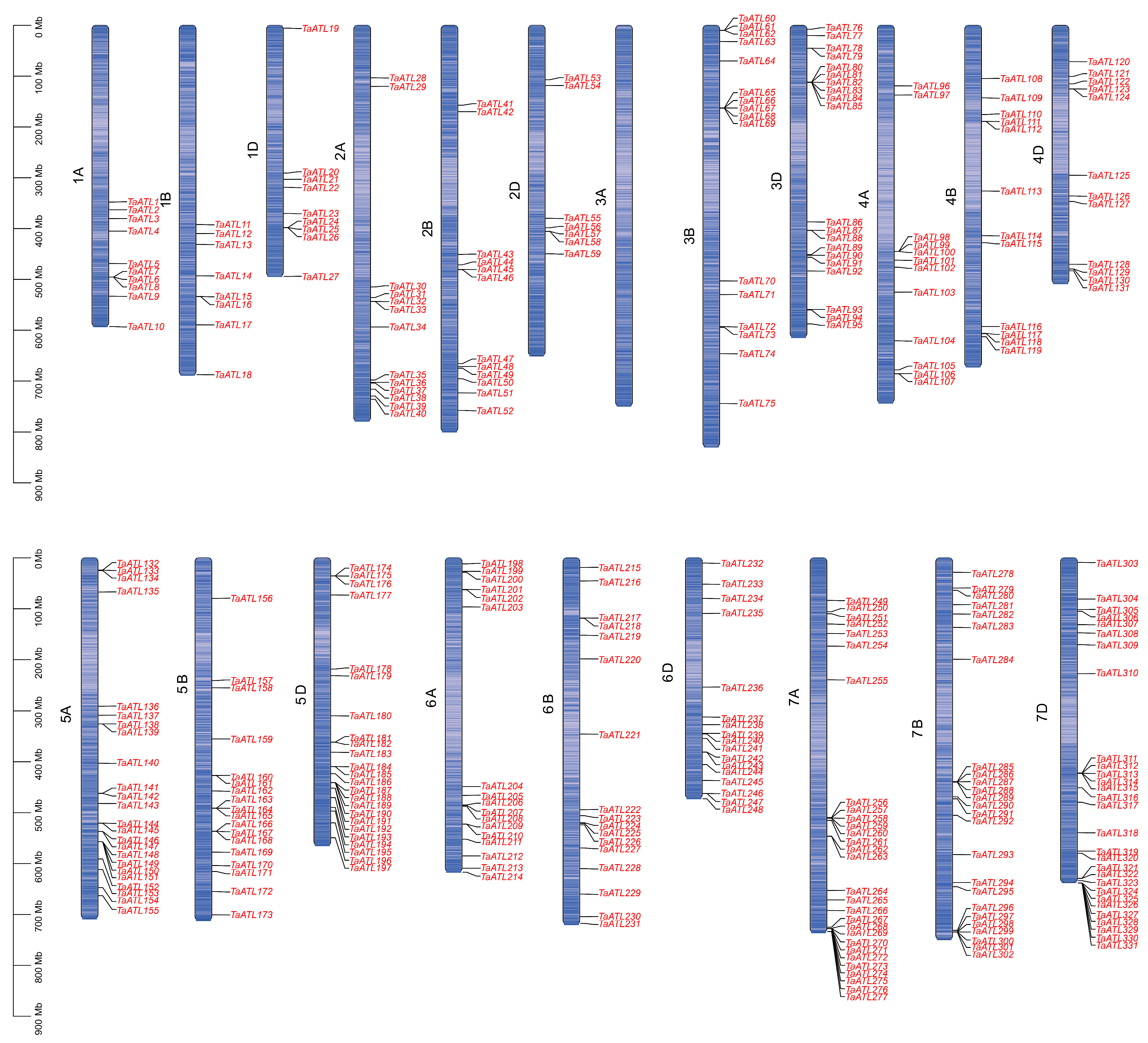
2.1. Genome-Wide Identification of TaATL Family Genes
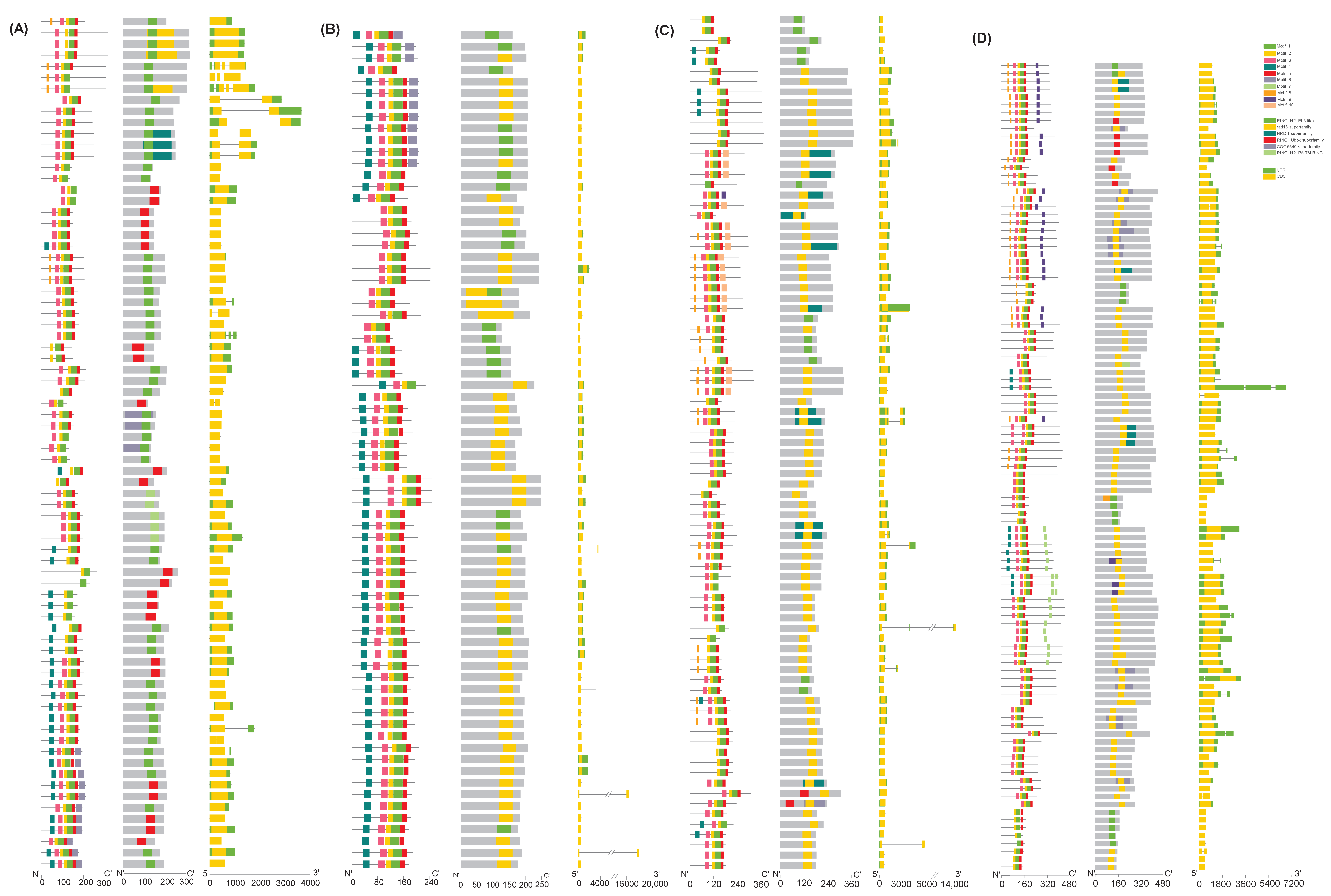
2.2. Gene Structure of TaATL Family Genes
2.3. Conserved Domains and Motif Distribution of TaATL Family Genes
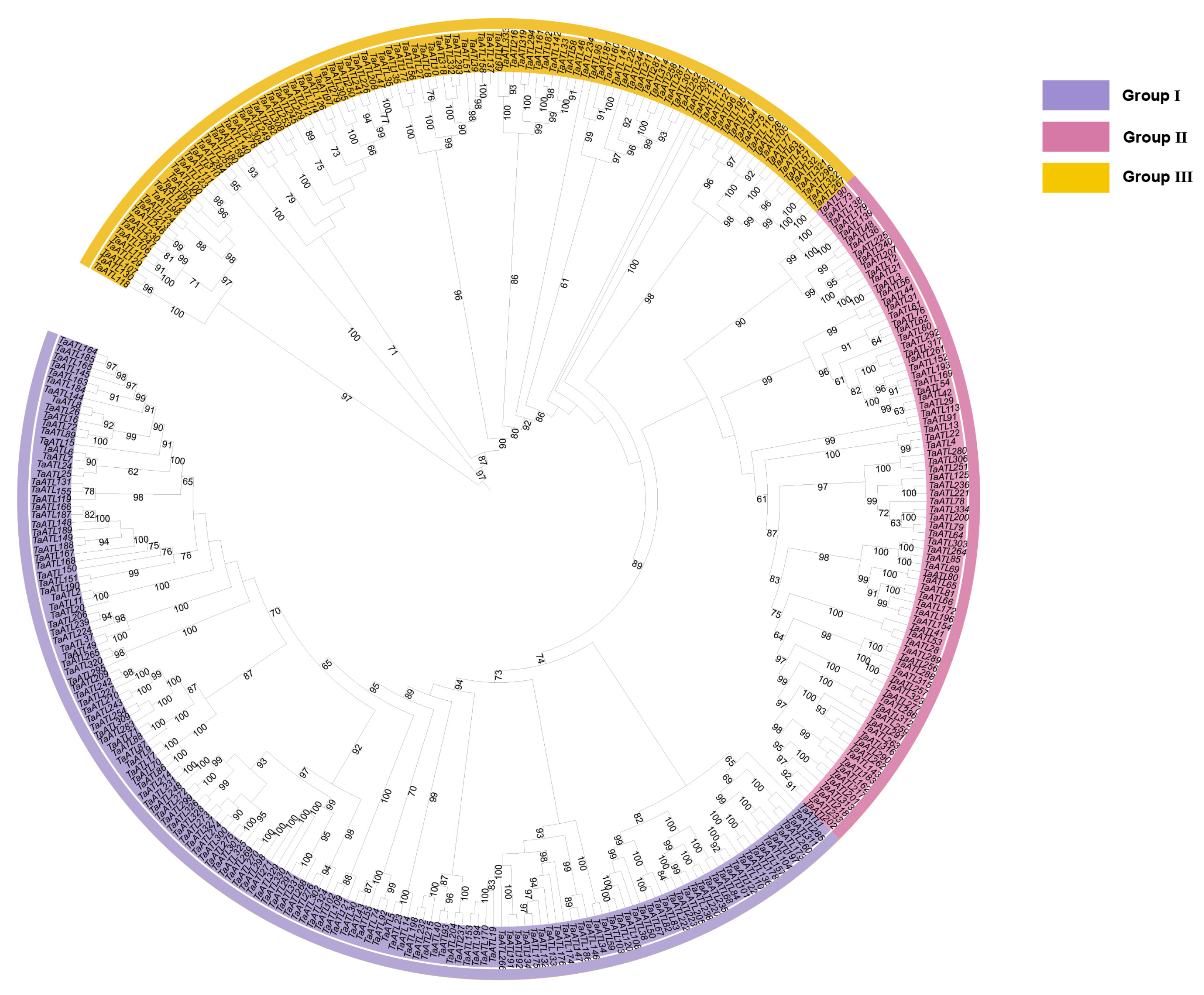
2.4. Gene Duplication Events and Orthology Analysis of TaATL Family Genes
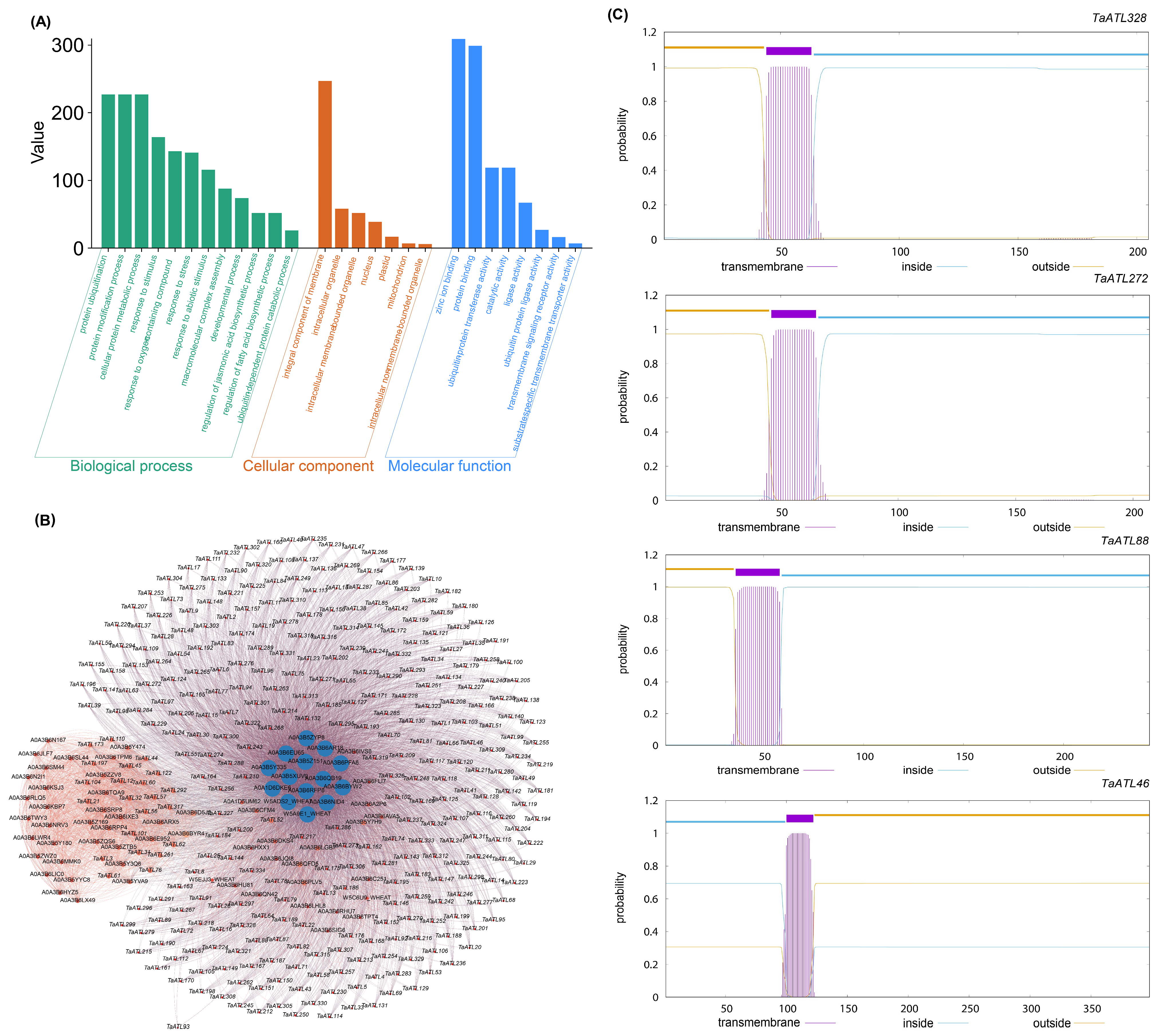
2.5. GO Analysis, Protein–Protein Interactions, and Transmembrane Domain Analysis of TaATL Family Genes
2.6. Cis-Regulatory Element Analysis of TaATL Genes
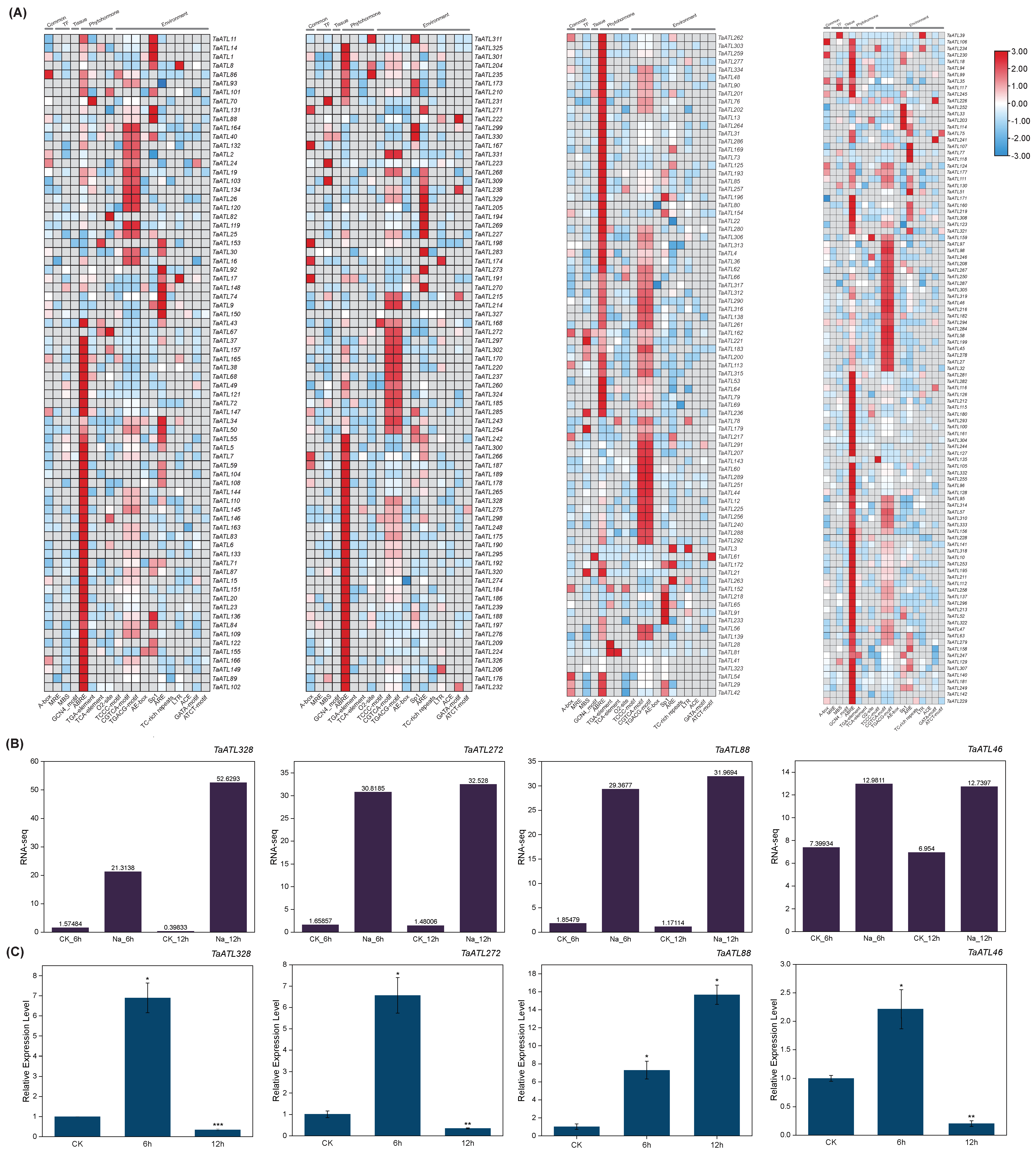
2.7. Analysis of Expression Levels of TaATL Family Genes Under Salt Stress
3. Discussion
4. Materials and Methods
4.1. Identification of TaATL Family Genes
4.2. Phylogenetic Analysis and Collinearity Analysis of TaATL Family Genes
4.3. Analysis of cis-Acting Elements, Gene Structure, and Chromosome Location
4.4. Protein–Protein Interaction, GO, and Physicochemical Property Analyses
4.5. Plant Materials and Salt Stress Treatment
4.6. Gene Expression Analysis, RNA Extraction, and Real-Time Quantitative PCR (RT-qPCR) Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATL | Arabidopsis Tóxicos en Levadura |
| RING | Really Interesting New Gene |
| RAD | RING-associated domain |
| ABA | Abscisic acid |
| MYB | Myeloblastosis |
| ABRE | ABA-responsive element |
| MeJA | Methyl jasmonate |
| MBS | MYB-binding site |
| UPS | Ubiquitin–proteasome system |
| UTR | Untranslated region |
| GO | Gene Ontology |
| Ka | Nonsynonymous substitution rate |
| Ks | Synonymous substitution rate |
| PPI | Protein–protein interaction |
References
- Bailey-Serres, J.; Parker, J.E.; Ainsworth, E.A.; Oldroyd, G.E.D.; Schroeder, J.I. Genetic Strategies for Improving Crop Yields. Nature 2019, 575, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Al-Saharin, R.; Hellmann, H.; Mooney, S. Plant E3 Ligases and Their Role in Abiotic Stress Response. Cells 2022, 11, 890. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, J.H.; Amoah, J.N.; Seo, Y.W. Wheat (Triticum aestivum L.) Plant U-box E3 Ligases TaPUB2 and TaPUB3 Enhance ABA Response and Salt Stress Resistance in Arabidopsis. FEBS Lett. 2022, 596, 3037–3050. [Google Scholar] [CrossRef]
- Jing, Z.; Liu, N.; Zhang, Z.; Hou, X. Research Progress on Plant Responses to Stress Combinations in the Context of Climate Change. Plants 2024, 13, 469. [Google Scholar] [CrossRef]
- Stone, S.L. Role of the Ubiquitin Proteasome System in Plant Response to Abiotic Stress. Int. Rev. Cell Mol. Biol. 2019, 343, 1. [Google Scholar] [CrossRef]
- Parveen, A.; Rahim, M.S.; Sharma, A.; Mishra, A.; Kumar, P.; Fandade, V.; Kumar, P.; Bhandawat, A.; Verma, S.K.; Roy, J. Genome-Wide Analysis of RING-Type E3 Ligase Family Identifies Potential Candidates Regulating High Amylose Starch Biosynthesis in Wheat (Triticum aestivum L.). Sci. Rep. 2021, 11, 11461. [Google Scholar] [CrossRef]
- Ariani, P.; Regaiolo, A.; Lovato, A.; Giorgetti, A.; Porceddu, A.; Camiolo, S.; Wong, D.; Castellarin, S.; Vandelle, E.; Polverari, A. Genome-Wide Characterisation and Expression Profile of the Grapevine ATL Ubiquitin Ligase Family Reveal Biotic and Abiotic Stress-Responsive and Development-Related Members. Sci. Rep. 2016, 6, 38260. [Google Scholar] [CrossRef]
- Serrano, M.; Parra, S.; Alcaraz, L.D.; Guzmán, P. The ATL Gene Family from Arabidopsis Thaliana and Oryza Sativa Comprises a Large Number of Putative Ubiquitin Ligases of the RING-H2 Type. J. Mol. Evol. 2006, 62, 434–445. [Google Scholar] [CrossRef]
- Ding, H.; Li, X.; Zhuge, S.; Du, J.; Wu, M.; Li, W.; Li, Y.; Ma, H.; Zhang, P.; Wang, X.; et al. Genome-Wide Identification and Functional Analysis of the Genes of the ATL Family in Maize during High-Temperature Stress in Maize. Genes 2024, 15, 1106. [Google Scholar] [CrossRef]
- Wang, S.; Lv, X.; Zhang, J.; Chen, D.; Chen, S.; Fan, G.; Ma, C.; Wang, Y. Roles of E3 Ubiquitin Ligases in Plant Responses to Abiotic Stresses. Int. Rev. Cell Mol. Biol. 2022, 23, 2308. [Google Scholar] [CrossRef]
- Moon, J.; Parry, G.; Estelle, M. The Ubiquitin-Proteasome Pathway and Plant Development. Plant Cell 2004, 16, 3181–3195. [Google Scholar] [CrossRef] [PubMed]
- Homology-Mediated Inter-Chromosomal Interactions in Hexaploid Wheat Lead to Specific Subgenome Territories Following Polyploidization and Introgression | Genome Biology | Full Text. Available online: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02225-7 (accessed on 24 February 2025).
- Buzuk, L.; Hellerschmied, D. Ubiquitin-Mediated Degradation at the Golgi Apparatus. Front. Mol. Biosci. 2023, 10, 1197921. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, P. The Prolific ATL Family of RING-H2 Ubiquitin Ligases. Plant Signal. Behav. 2012, 7, 1014–1021. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One Step at a Time: Endoplasmic Reticulum-Associated Degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Reyes-Impellizzeri, S.; Moreno, A.A. The Endoplasmic Reticulum Role in the Plant Response to Abiotic Stress. Front. Plant Sci. 2021, 12, 755447. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of Cis-Acting Regulatory Elements in Osmotic- and Cold-Stress-Responsive Promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamaguchi-Shinozaki, K. ABA Signaling in Stress-Response and Seed Development. Plant Cell Rep. 2013, 32, 959–970. [Google Scholar] [CrossRef]
- Waadt, R.; Seller, C.A.; Hsu, P.-K.; Takahashi, Y.; Munemasa, S.; Schroeder, J.I. Plant Hormone Regulation of Abiotic Stress Responses. Nat. Rev. Mol. Cell Biol. 2022, 23, 680–694. [Google Scholar] [CrossRef]
- Wang, N.; Qu, C.; Wang, Y.; Xu, H.; Jiang, S.; Fang, H.; Liu, J.; Zhang, Z.; Chen, X. MdMYB4 Enhances Apple Callus Salt Tolerance by Increasing MdNHX1 Expression Levels. Plant Cell Tissue Organ Cult. PCTOC 2017, 131, 283–293. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB Transcription Factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Wang, X.; Wei, H.; Wang, K.; Tang, X.; Li, S.; Zhang, N.; Si, H. MYB Transcription Factors: Acting as Molecular Switches to Regulate Different Signaling Pathways to Modulate Plant Responses to Drought Stress. Ind. Crops Prod. 2025, 226, 120676. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional Control of Flavonoid Biosynthesis by MYB–bHLH–WDR Complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Raskin, I. Role of Salicylic Acid in Plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1992, 43, 439–463. [Google Scholar] [CrossRef]
- Ramírez, V.; Coego, A.; López, A.; Agorio, A.; Flors, V.; Vera, P. Drought Tolerance in Arabidopsis Is Controlled by the OCP3 Disease Resistance Regulator. Plant J. 2009, 58, 578–591. [Google Scholar] [CrossRef]
- Miura, K.; Tada, Y. Regulation of Water, Salinity, and Cold Stress Responses by Salicylic Acid. Front. Plant Sci. 2014, 5, 4. [Google Scholar] [CrossRef]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C Phosphatase Activity Function as Abscisic Acid Sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef]
- Santiago, J.; Dupeux, F.; Betz, K.; Antoni, R.; Gonzalez-Guzman, M.; Rodriguez, L.; Márquez, J.A.; Rodriguez, P.L. Structural Insights into PYR/PYL/RCAR ABA Receptors and PP2Cs. Plant Sci. 2012, 182, 3–11. [Google Scholar] [CrossRef]
- Fahmideh, L.; Fooladvand, Z. Isolation and Semi Quantitative PCR of Na+/H+ Antiporter (SOS1 and NHX) Genes under Salinity Stress in Kochia Scoparia. Biol. Proced. Online 2018, 20, 11. [Google Scholar] [CrossRef]
- Li, W.; Wei, J.; Lei, Y.; Yang, Z.; Zhang, S.; Feng, J.; Li, Y.; Liu, Y.; Sheng, H. Phosphorylation of Cellulose Synthases in Plant Responses to Environmental Changes. Int. J. Biol. Macromol. 2025, 292, 139313. [Google Scholar] [CrossRef]
- Bolser, D.M.; Staines, D.M.; Perry, E.; Kersey, P.J. Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomic Data. Methods Mol. Biol. 2017, 1533, 1–31. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “One for All, All for One” Bioinformatics Platform for Biological Big-Data Mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: The Protein Sequence Classification Resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era | Molecular Biology and Evolution | Oxford Academic. Available online: https://academic.oup.com/mbe/article/37/5/1530/5721363 (accessed on 1 March 2025).
- Balister, P.; Bollobás, B.; Przykucki, M.; Smith, P. Subcritical -Bootstrap Percolation Models Have Non-Trivial Phase Transitions. Trans. Am. Math. Soc. 2016, 368, 7385–7411. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Chen, K.; Durand, D.; Farach-Colton, M. NOTUNG: A Program for Dating Gene Duplications and Optimizing Gene Family Trees. J. Comput. Biol. 2000, 7, 429–447. [Google Scholar] [CrossRef]
- RStudio: Integrated Development for R. Available online: http://www.rstudio.com/ (accessed on 12 December 2024).
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Déhais, P.; Van Montagu, M.; Rouzé, P. PlantCARE, a Plant Cis-Acting Regulatory Element Database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Panzade, G.; Imamichi, T.; Chang, W. DAVID Ortholog: An Integrative Tool to Enhance Functional Analysis through Orthologs. Bioinformatics 2024, 40, btae615. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. ISBN 978-1-58829-343-5. [Google Scholar]
- Yu, C.-S.; Lin, C.-J.; Hwang, J.-K. Predicting Subcellular Localization of Proteins for Gram-Negative Bacteria by Support Vector Machines Based on n-Peptide Compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive Protein Feature Visualization and Integration with Experimental Proteomic Data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef]
- Ying, S. Genome-Wide Identification and Transcriptional Analysis of Arabidopsis DUF506 Gene Family. Int. J. Mol. Sci. 2021, 22, 11442. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Ma, S.; Wang, M.; Wu, J.; Guo, W.; Chen, Y.; Li, G.; Wang, Y.; Shi, W.; Xia, G.; Fu, D.; et al. WheatOmics: A Platform Combining Multiple Omics Data to Accelerate Functional Genomics Studies in Wheat. Mol. Plant 2021, 14, 1965–1968. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Son, S.; Chitnis, V.R.; Liu, A.; Gao, F.; Nguyen, T.-N.; Ayele, B.T. Abscisic Acid Metabolic Genes of Wheat (Triticum aestivum L.): Identification and Insights into Their Functionality in Seed Dormancy and Dehydration Tolerance. Planta 2016, 244, 429–447. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, L.; Zhao, W.; Fu, L.; Han, Y.; Wang, K.; Yan, L.; Li, Y.; Zhang, X.-H.; Min, D.-H. Genome-Wide Analysis of the Serine Carboxypeptidase-like Protein Family in Triticum aestivum Reveals TaSCPL184-6D Is Involved in Abiotic Stress Response. BMC Genom. 2021, 22, 350. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Shen, S.; Wang, Y.; Tao, W.; Zhao, Z.; Hu, X.; Yu, P. Genome-Wide Identification and Expression Analysis of the Zinc Finger Protein Gene Subfamilies under Drought Stress in Triticum aestivum. Plants 2022, 11, 2511. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, S.; Wu, Z.; Tao, W.; Zhang, W.; Yu, P. Genome-Wide Analysis of Triticum aestivum Bromodomain Gene Family and Expression Analysis under Salt Stress. Planta 2024, 260, 117. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Liu, S.; Yu, P. Genome-Wide Identification of ATL Gene Family in Wheat and Their Expression Analysis in Response to Salt Stress. Plants 2025, 14, 1306. https://doi.org/10.3390/plants14091306
Li X, Liu S, Yu P. Genome-Wide Identification of ATL Gene Family in Wheat and Their Expression Analysis in Response to Salt Stress. Plants. 2025; 14(9):1306. https://doi.org/10.3390/plants14091306
Chicago/Turabian StyleLi, Xuqing, Shuotong Liu, and Pei Yu. 2025. "Genome-Wide Identification of ATL Gene Family in Wheat and Their Expression Analysis in Response to Salt Stress" Plants 14, no. 9: 1306. https://doi.org/10.3390/plants14091306
APA StyleLi, X., Liu, S., & Yu, P. (2025). Genome-Wide Identification of ATL Gene Family in Wheat and Their Expression Analysis in Response to Salt Stress. Plants, 14(9), 1306. https://doi.org/10.3390/plants14091306