

Development and Application of SSR Markers for Aquilaria sinensis on the Basis of Whole-Genome Resequencing Data

 , and
, and
Abstract
1. Introduction
2. Results
2.1. SSR Locus Characteristics
2.2. SSR Primer Development
2.3. SSR Polymorphism Evaluation
2.4. Genetic Diversity Analysis
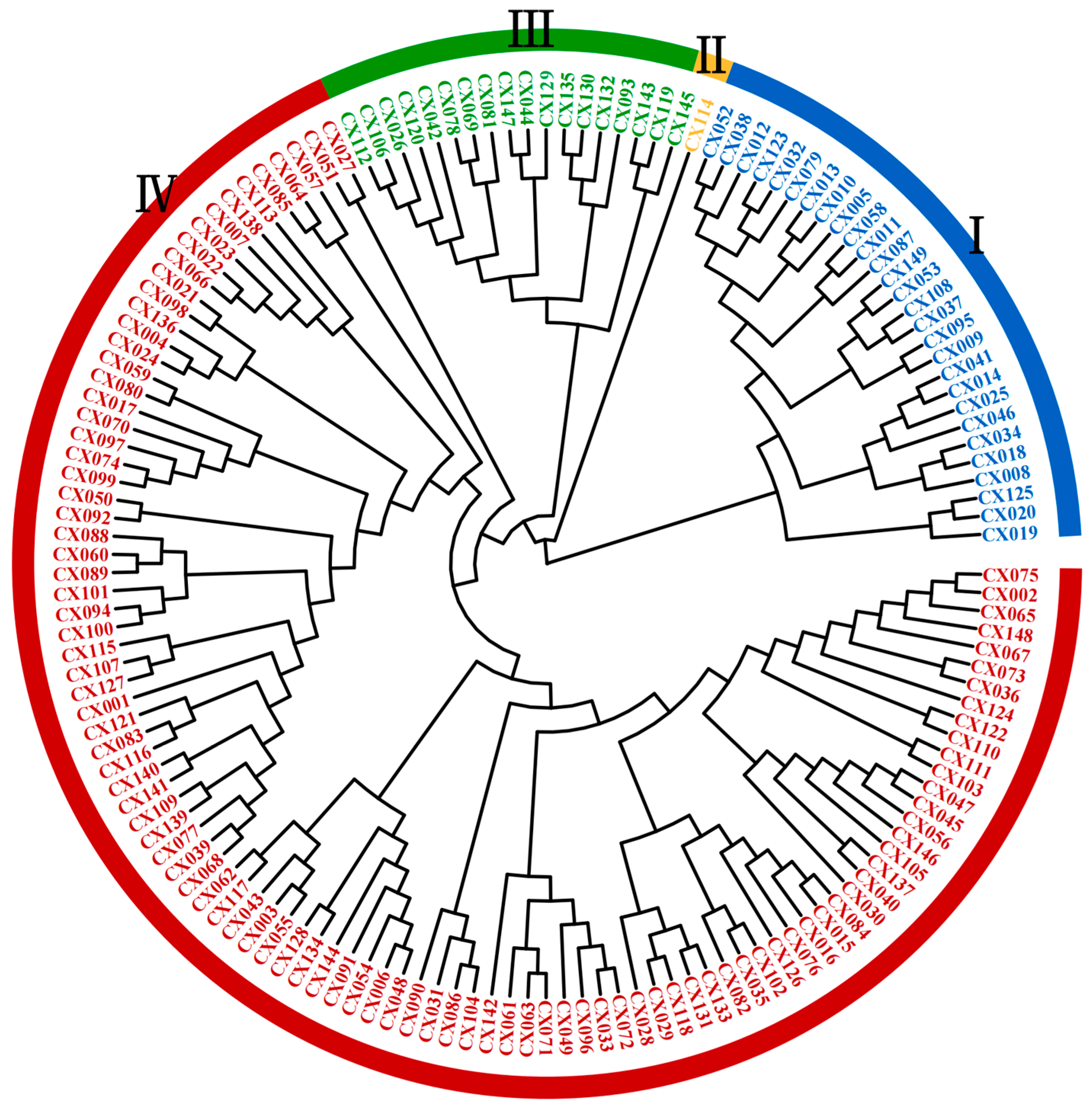
2.4.1. Clustering Analysis
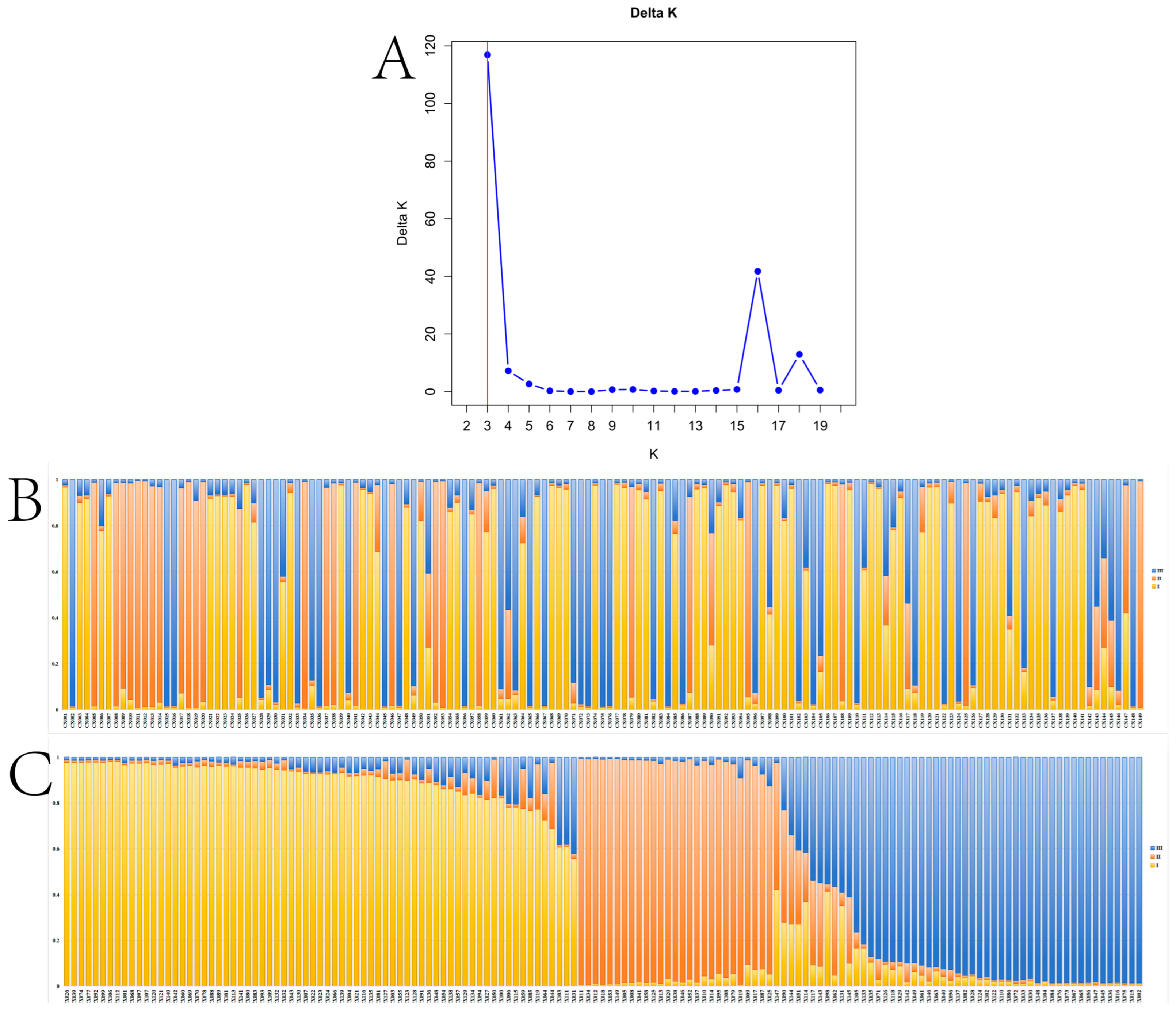
2.4.2. Population Genetic Structure
2.4.3. Population Genetic Diversity Analysis
2.4.4. Fingerprint Profiling
3. Discussion
4. Materials and Methods
4.1. DNA Extraction
4.2. SSR Locus Identification and Primer Design
4.3. SSR-Based Genotyping
4.4. SSR Polymorphism and Genetic Diversity Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mohamed, R.; Jong, P.L.; Kamziah, A.K. Fungal inoculation induces agarwood in young Aquilaria malaccensis trees in the nursery. J. For. Res. 2014, 25, 201–204. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, H.; Lyu, F.; Fan, X.; Liu, P.; He, X.; Huang, Y.; Chen, B.; Yang, Y.; Wei, J. Temporal characteristics of agarwood formation in Aquilaria sinensis after applying whole-tree agarwood-inducing technique. Chin. Herb. Med. 2023, 15, 37–44. [Google Scholar] [CrossRef]
- Wang, B.; Ma, G.; Lin, S.; He, X.; Chen, B.; Li, H.; Huang, L.; Yang, Y.; Wei, J. Floral Biology of Aquilaria sinensis (Lour.) Spreng. Hortic. 2024, 10, 109. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C.; Sun, W. Pollination and seed dispersal of Aquilaria sinensis (Lour.) Gilg (Thymelaeaceae): An economic plant species with extremely small populations in China. Plant Divers. 2016, 38, 227–232. [Google Scholar] [CrossRef]
- Lopez-Sampson, A.; Page, T. History of Use and Trade of Agarwood. Econ. Bot. 2018, 72, 107–129. [Google Scholar] [CrossRef]
- Dahham, S.S.; Hassan, L.E.A.; Ahamed, M.B.K.; Majid, A.S.A.; Majid, A.M.S.A.; Zulkepli, N.N. In vivo toxicity and antitumor activity of essential oils extract from agarwood (Aquilaria crassna). Bmc Complement. Altern. Med. 2016, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, Y.; Xue, J.; Wei, J.; Zhang, Z.; Chen, H. Comparison of Compositions and Antimicrobial Activities of Essential Oils from Chemically Stimulated Agarwood, Wild Agarwood and Healthy Aquilaria sinensis (Lour.) Gilg Trees. Molecules 2011, 16, 4884–4896. [Google Scholar] [CrossRef]
- Li, W.; Chen, H.-Q.; Wang, H.; Mei, W.-L.; Dai, H.-F. Natural products in agarwood and Aquilaria plants: Chemistry, biological activities and biosynthesis. Nat. Prod. Rep. 2021, 38, 528–565. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.; Peng, D.; Liu, X.; Wu, C.; Guo, P.; Wei, J. Agarwood Essential Oil Displays Sedative-Hypnotic Effects through the GABAergic System. Molecules 2017, 22, 2190. [Google Scholar] [CrossRef]
- Xie, C.; Dong, J.-Z.; Lu, B.-S.; Yan, P.-Y.; Zhao, Y.-S.; Ding, X.-Y.; Lv, C.-E.; Zheng, X. Pharmacology and therapeutic potential of agarwood and agarwood tree leaves in periodontitis. Front. Pharmacol. 2024, 15, 1468393. [Google Scholar] [CrossRef]
- Yu, Z.-X.; Wang, C.-H.; Chen, D.-L.; Liu, Y.-Y.; Wei, J.-H. Anti-inflammatory sesquiterpenes from agarwood produced via whole-tree agarwood-inducing technique of Aquilaria sinensis. China J. Chin. Mater. Medica 2019, 44, 4196–4202. [Google Scholar]
- Akter, S.; Islam, M.; Zusoh, Z.; Khan, S. Agarwood production-a multidisciplinary field to be explored in Bangla-desh. Int. J. Pharm. Life Sci. 2013, 2, 22–32. [Google Scholar] [CrossRef]
- Xu, J.; Du, R.; Wu, K.; Chen, J. Development of SSR markers related to agarwood production and genetic diversity of Aquilaria sinensis (Lour.) Spreng wild populations. J. Appl. Res. Med. Aromat. Plants 2024, 42, 100565. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Yang, Y.; Zhang, Z.; Wei, J.; Meng, H.; Chen, W.; Feng, J.; Gan, B.; Chen, X.; et al. Whole-tree Agarwood-Inducing Technique: An Efficient Novel Technique for Producing High-Quality Agarwood in Cultivated Aquilaria sinensis Trees. Molecules 2013, 18, 3086–3106. [Google Scholar] [CrossRef]
- Ma, S.; Fu, Y.; Li, Y.; Wei, P.; Liu, Z. The formation and quality evaluation of agarwood induced by the fungi in Aquilaria sinensis. Ind. Crops Prod. 2021, 173, 114129. [Google Scholar] [CrossRef]
- Tan, C.S.; Isa, N.M.; Ismail, I.; Zainal, Z. Agarwood Induction: Current Developments and Future Perspectives. Front. Plant Sci. 2019, 10, 122. [Google Scholar] [CrossRef]
- Sraphet, S.; Boonchanawiwat, A.; Thanyasiriwat, T.; Boonseng, O.; Tabata, S.; Sasamoto, S.; Shirasawa, K.; Isobe, S.; Lightfoot, D.A.; Tangphatsornruang, S.; et al. SSR and EST-SSR-based genetic linkage map of cassava (Manihot esculenta Crantz). Theor. Appl. Genet. 2011, 122, 1161–1170. [Google Scholar] [CrossRef]
- Wu, F.; Cai, G.; Xi, P.; Guo, Y.; Xu, M.; Li, A. Genetic Diversity Analysis and Fingerprint Construction for 87 Passionfruit (Passiflora spp.) Germplasm Accessions on the Basis of SSR Fluorescence Markers. Int. J. Mol. Sci. 2024, 25, 10815. [Google Scholar] [CrossRef]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (ssr) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef]
- McCouch, S.R.; Teytelman, L.; Xu, Y.; Lobos, K.B.; Clare, K.; Walton, M.; Fu, B.; Maghirang, R.; Li, Z.; Xing, Y.; et al. Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2002, 9, 199–207. [Google Scholar]
- Tyagi, S.; Kumar, A.; Gautam, T.; Pandey, R.; Rustgi, S.; Mir, R.R. Development and use of miRNA-derived SSR markers for the study of genetic diversity, population structure, and characterization of genotypes for breeding heat tolerant wheat varieties. PLoS ONE 2021, 16, e0231063. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Xia, Z.; Lu, C.; Wang, H.; Ji, J.; Wang, W. Genetic Diversity and Differentiation of Aquilaria sinensis (Lour.) Gilg Revealed by ISSR and SRAP Markers. Crop Sci. 2012, 52, 2304–2313. [Google Scholar] [CrossRef]
- Song, B.; Ning, W.; Wei, D.; Jiang, M.; Zhu, K.; Wang, X.; Edwards, D.; Odeny, D.A.; Cheng, S. Plant genome resequencing and population genomics: Current status and future prospects. Mol. Plant 2023, 16, 1252–1268. [Google Scholar] [CrossRef]
- Ouni, R.; Zborowska, A.; Sehic, J.; Choulak, S.; Inaki Hormaza, J.; Garkava-Gustavsson, L.; Mars, M. Genetic Diversity and Structure of Tunisian Local Pear Germplasm as Revealed by SSR Markers. Hortic. Plant J. 2020, 6, 61–70. [Google Scholar] [CrossRef]
- Meng, Y.; Zheng, C.; Li, H.; Li, A.; Zhai, H.; Wang, Q.; He, S.; Zhao, N.; Zhang, H.; Gao, S.; et al. Development of a high-density SSR genetic linkage map in sweet potato. Crop J. 2021, 9, 1367–1374. [Google Scholar] [CrossRef]
- Yan, X.C.; Ma, X.; Lan, J.H. Genetic diversity analysis of maize germplasm resources using SSR molecular markers. J. Qingdao Agric. Univ. (Ziran Kexueban) 2024, 41, 1–9. [Google Scholar]
- Li, J.; Li, S.; Kong, L.; Wang, L.; Wei, A.; Liu, Y. Genome survey of Zanthoxylum bungeanum and development of genomic-SSR markers in congeneric species. Biosci. Rep. 2020, 40, BSR20201101. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Wu, K.; Chen, J. Identification and characterization of functionally relevant SSR markers in natural Dalbergia odorifera populations. Bmc Plant Biol. 2024, 24, 315. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, D.F.; Fan, G.H.; Wang, Z.L. Comparative analysis of SSR distribution characteristics between transcriptome and genome of Lycium barbarum. Jiangsu Agric. Sci. 2018, 46, 24–27. [Google Scholar]
- Wang, Z.F.; Cao, H.L.; Cai, C.-X.; Wang, Z.-M. Using genetic markers to identify the origin of illegally traded agarwood-producing Aquilaria sinensis trees. Glob. Ecol. Conserv. 2020, 22, e00958. [Google Scholar] [CrossRef]
- Gou, Z.H.; Chen, G.D.; Zhen, Y.N.; Wang, X.; Chen, Y.; Tian, M. Genetic diversity of Aquilaria sinensis based on genome resequencing. Chin. J. Trop. Crops 2025, 46, 51–58. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2025; Available online: https://www.R-project.org (accessed on 5 August 2024).

{kind=link}
{kind=link}
| SSR Locus Characteristics | Quantity |
|---|---|
| Total number of identified sequences | 168,459 |
| SSR sequences | 56,657 |
| SSR loci | 24,430 |
| Mononucleotide repeats | 4747 |
| Dinucleotide repeats | 41,695 |
| Trinucleotide repeats | 9550 |
| Tetranucleotide repeats | 644 |
| Pentanucleotide repeats | 13 |
| Hexanucleotide repeats | 8 |
| SSR Primer | SSR Locus ID | Motif | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|---|---|
| AquSSR07 | Scaffold_5532_102997041 | CA | AAACGGAACGTGCTAATGCT | CCACACGTGATTTCTGGATG |
| AquSSR10 | Scaffold_9996_84841276 | ATG | TATTAGTGGGTGAATCGGGC | GGGCAAACGGTATAATCATCA |
| AquSSR14 | Scaffold_15334_39078260 | AT | GACATAAGGGGCCATGAGTG | AAGCCTAGCCTTTTTGGTGG |
| AquSSR17 | Scaffold_10796_83936585 | AAT | CAAAACCAAATTCACTTGAAAGC | CCACCAGCACAAGTGGTATG |
| AquSSR18 | Scaffold_10796_86795031 | ATT | GGAGAGGGTTGAGGTAGGGT | CGGTGTTTGAGATTGTGGAA |
| AquSSR22 | Scaffold_8152_21061737 | TG | GGAACTCAATAGGCTGCTGG | CAAATTTTGGGTTGGGTACG |
| AquSSR27 | Scaffold_3585_12180739 | TTC | CATTTTACTTTTTGGCGGGA | TGCAACACAAGCAACACAAA |
| AquSSR28 | Scaffold_3585_77017729 | AAT | CGAGTGAGGGTTCACCAACT | TGCTCCATAAATGCATGCTC |
| AquSSR29 | Scaffold_10546_3054275 | GAA | AACACCTTCATCACCGGAAG | GGGCTTTTGTCATTTTCCCT |
| AquSSR30 | Scaffold_10546_7777087 | GA | TTAGCATGGTTTTGTGCTGG | TGCACAACCTCCTCTCTGTG |
| AquSSR34 | Scaffold_10546_70298677 | ATAC | ACCATGGACCACAGAGAAGC | AAGGGTATGTGTTGAAGGCG |
| AquSSR40 | Scaffold_10433_55273834 | TTA | TCTCCCACGTTTCCAACTTC | TTTGGTCACGAAAAGTGGTG |
| AquSSR42 | Scaffold_10433_59579622 | TAT | AACCCTTGTTTGAATGCAGG | CCTAATGGCTGAAAGCCTGA |
| AquSSR54 | Scaffold_10796_47706258 | TTA | TGCCCTTTAGACCATGGAAG | AGACCAATAGACCCAAGATGG |
| AquSSR58 | Scaffold_3585_1832452 | AAC | CAATGGGGTTTCTACAGGCA | TTGTTGGACATCACAAACGG |
| AquSSR59 | Scaffold_3585_64368533 | GCT | AGGGGAGGTGAAGAAAAGGA | CCATAACCATAGCAGCAGCA |
| AquSSR62 | Scaffold_10546_2555776 | ATA | TGTGTGGGTAAAATGAAGGCT | TGCCTAAATCTCCTTTGCTTTC |
| AquSSR71 | Scaffold_5532_21554441 | AAG | CGCAACCTCATGGGTAACTT | AACCAATCCTCAAACCTCCC |
| AquSSR89 | Scaffold_10546_79030278 | TAA | TTTTAATCAGGGGAGGACCC | TCTGCTGACGTGTACGGTTC |
| AquSSR94 | Scaffold_10433_72319168 | TAA | CCACTGTTTCTGCAAGCTAGG | GACTTCGTGATCTCAACGGG |
| Locus | N | Na | Ne | I | Ho | He | F | PIC |
|---|---|---|---|---|---|---|---|---|
| AquSSR07 | 149 | 3 | 2.014 | 0.820 | 0.423 | 0.503 | 0.160 | 0.422 |
| AquSSR10 | 149 | 6 | 3.751 | 1.403 | 0.725 | 0.733 | 0.012 | 0.688 |
| AquSSR14 | 149 | 6 | 1.256 | 0.478 | 0.195 | 0.204 | 0.047 | 0.196 |
| AquSSR17 | 149 | 4 | 1.410 | 0.578 | 0.282 | 0.291 | 0.031 | 0.268 |
| AquSSR18 | 149 | 10 | 2.049 | 1.168 | 0.389 | 0.512 | 0.240 | 0.489 |
| AquSSR22 | 149 | 4 | 3.048 | 1.193 | 0.181 | 0.672 | 0.730 | 0.608 |
| AquSSR27 | 149 | 7 | 3.599 | 1.456 | 0.497 | 0.722 | 0.312 | 0.676 |
| AquSSR28 | 149 | 6 | 1.891 | 0.963 | 0.389 | 0.471 | 0.174 | 0.443 |
| AquSSR29 | 149 | 12 | 2.496 | 1.282 | 0.671 | 0.599 | −0.120 | 0.564 |
| AquSSR30 | 149 | 7 | 3.148 | 1.236 | 0.564 | 0.682 | 0.174 | 0.617 |
| AquSSR34 | 148 | 3 | 1.344 | 0.493 | 0.250 | 0.256 | 0.023 | 0.236 |
| AquSSR40 | 148 | 7 | 3.308 | 1.457 | 0.615 | 0.698 | 0.119 | 0.660 |
| AquSSR42 | 148 | 5 | 2.904 | 1.192 | 0.581 | 0.656 | 0.114 | 0.595 |
| AquSSR54 | 149 | 9 | 2.680 | 1.221 | 0.631 | 0.627 | −0.006 | 0.564 |
| AquSSR58 | 149 | 5 | 2.074 | 0.864 | 0.477 | 0.518 | 0.080 | 0.434 |
| AquSSR59 | 148 | 5 | 3.706 | 1.365 | 0.716 | 0.730 | 0.019 | 0.680 |
| AquSSR62 | 149 | 6 | 2.627 | 1.098 | 0.550 | 0.619 | 0.112 | 0.554 |
| AquSSR71 | 148 | 5 | 1.939 | 0.832 | 0.405 | 0.484 | 0.163 | 0.410 |
| AquSSR89 | 145 | 3 | 1.964 | 0.781 | 0.428 | 0.491 | 0.129 | 0.402 |
| AquSSR94 | 147 | 8 | 3.009 | 1.389 | 0.932 | 0.668 | −0.396 | 0.627 |
| Mean | 148.45 | 6.050 | 2.511 | 1.063 | 0.495 | 0.557 | 0.106 | 0.507 |
| Population | N | Na | Ne | I | Ho | He | F |
|---|---|---|---|---|---|---|---|
| I | 70.750 | 4.600 | 2.280 | 0.934 | 0.484 | 0.505 | 0.050 |
| II | 29.750 | 4.800 | 2.816 | 1.141 | 0.561 | 0.603 | 0.063 |
| III | 47.950 | 3.850 | 2.141 | 0.851 | 0.471 | 0.470 | −0.002 |
| Mean | 49.483 | 4.417 | 2.412 | 0.975 | 0.505 | 0.526 | 0.037 |
| Source of Variation | Degrees of Freedom (df) | Sum of Squares (SS) | Mean Squares (MS) | Estimated Variance | Percentage of Variation (%) |
|---|---|---|---|---|---|
| Among Populations | 2 | 130.156 | 65.078 | 0.636 | 11% |
| Within Populations | 146 | 804.267 | 5.509 | 0.290 | 5% |
| Among Individuals | 149 | 734.500 | 4.930 | 4.930 | 84% |
| Total | 297 | 1668.923 | 5.855 | 100% |
| Population | Ⅰ | Ⅱ | Ⅲ |
|---|---|---|---|
| I | 0.000 | ||
| II | 0.056 | 0.000 | |
| III | 0.051 | 0.085 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Wu, K.; Xu, J.; Zhao, S.; Tu, Z.; Rao, D.; Chen, B.; Jiao, N.; Chen, J.; Dong, X. Development and Application of SSR Markers for Aquilaria sinensis on the Basis of Whole-Genome Resequencing Data. Plants 2025, 14, 1323. https://doi.org/10.3390/plants14091323
Chen Y, Wu K, Xu J, Zhao S, Tu Z, Rao D, Chen B, Jiao N, Chen J, Dong X. Development and Application of SSR Markers for Aquilaria sinensis on the Basis of Whole-Genome Resequencing Data. Plants. 2025; 14(9):1323. https://doi.org/10.3390/plants14091323
Chicago/Turabian StyleChen, Yu, Kunlin Wu, Jieru Xu, Shenghe Zhao, Zhihua Tu, Dandan Rao, Beibei Chen, Nanbo Jiao, Jinhui Chen, and Xiaona Dong. 2025. "Development and Application of SSR Markers for Aquilaria sinensis on the Basis of Whole-Genome Resequencing Data" Plants 14, no. 9: 1323. https://doi.org/10.3390/plants14091323
APA StyleChen, Y., Wu, K., Xu, J., Zhao, S., Tu, Z., Rao, D., Chen, B., Jiao, N., Chen, J., & Dong, X. (2025). Development and Application of SSR Markers for Aquilaria sinensis on the Basis of Whole-Genome Resequencing Data. Plants, 14(9), 1323. https://doi.org/10.3390/plants14091323