


Smoothness of Graph Energy in Chemical Graphs


Abstract
:1. Introduction
2. Preliminaries
3. Computational Details and Results
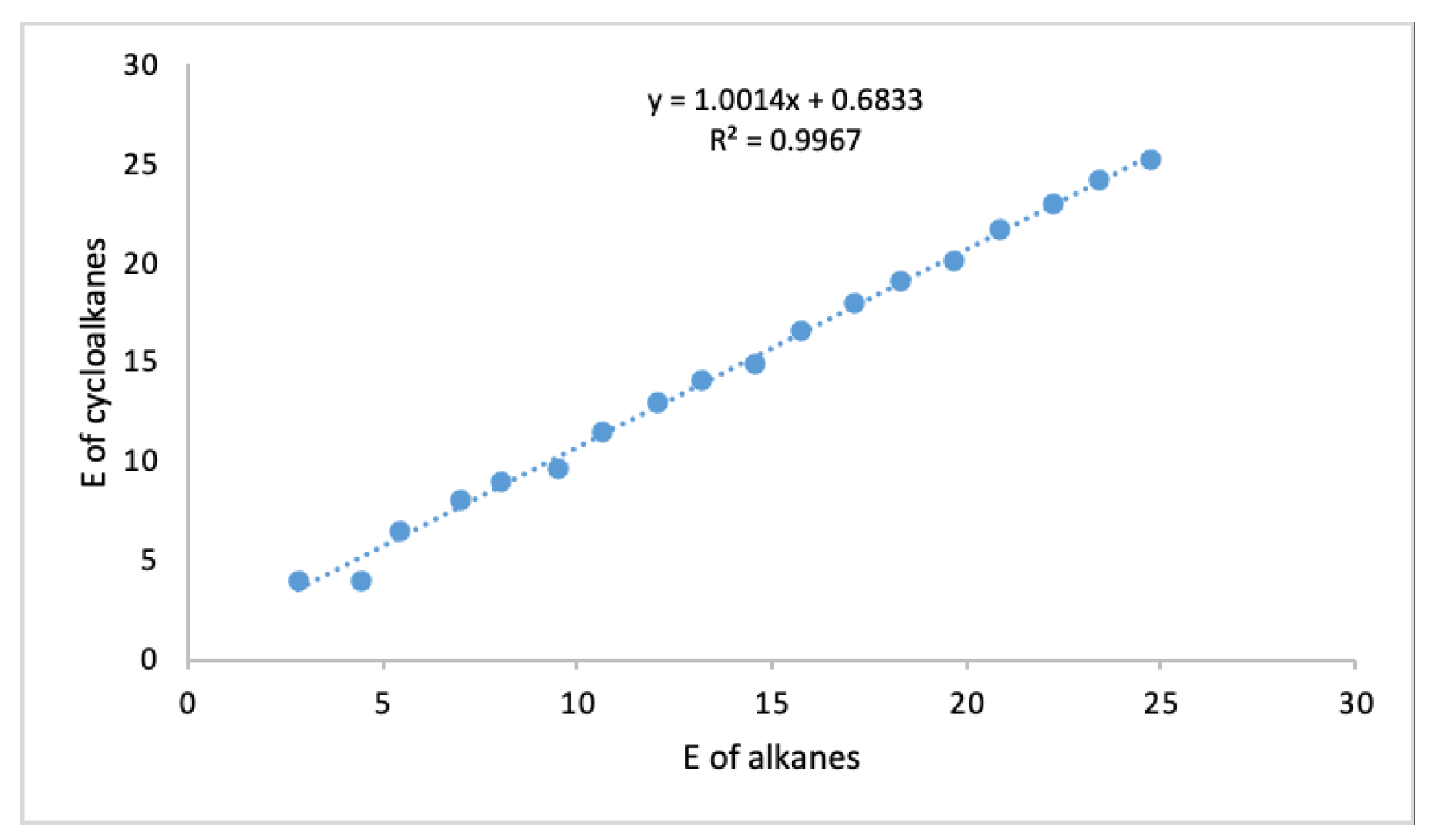
3.1. Linear Regression Analysis of Graph Energy and Correlations
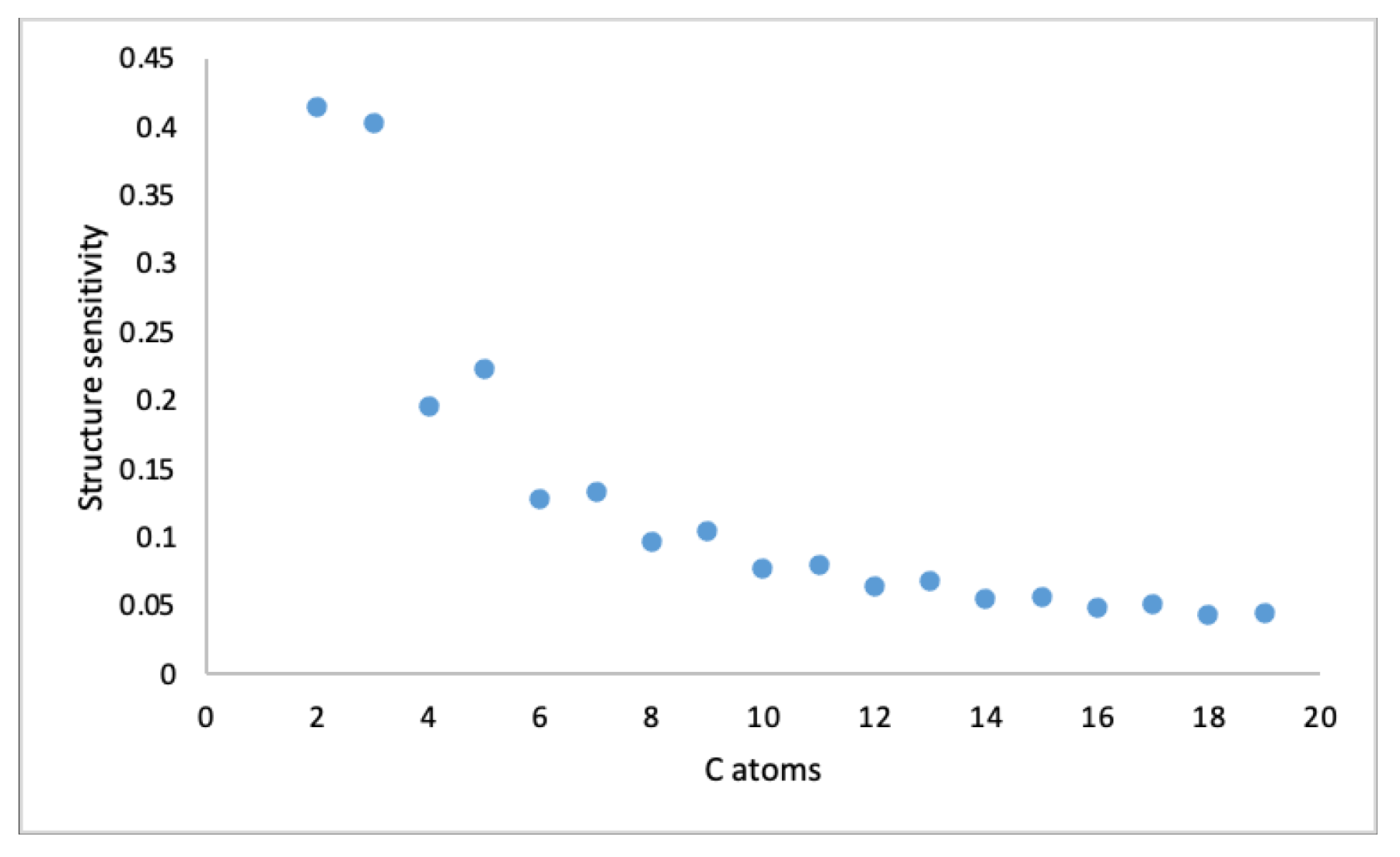
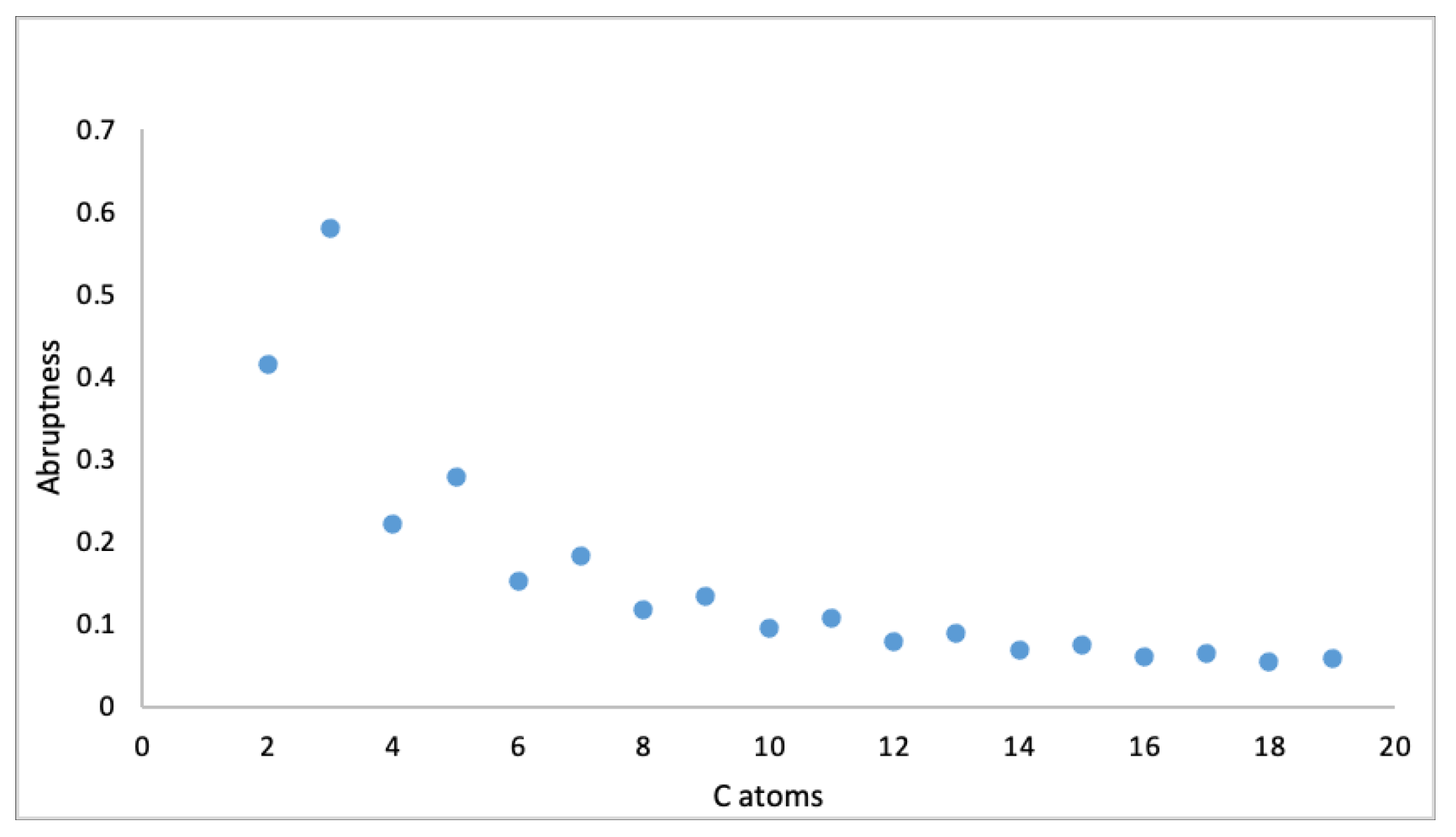
3.2. Structure Sensitivity and Abruptness of Graph Energy
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. Supplementary Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G | |||
|---|---|---|---|
| ethane | 1 | 0.41421 | 0.41421 |
| propane | 2 | 0.40294 | 0.58114 |
| butane | 2 | 0.19522 | 0.22181 |
| pentane | 3 | 0.22266 | 0.27888 |
| hexane | 3 | 0.12864 | 0.15266 |
| heptane | 4 | 0.13343 | 0.18162 |
| octane | 4 | 0.09627 | 0.11662 |
| nonane | 5 | 0.10433 | 0.13417 |
| decane | 5 | 0.07706 | 0.09443 |
| undecane | 6 | 0.07971 | 0.10620 |
| dodecane | 6 | 0.06432 | 0.07936 |
| tridecane | 7 | 0.06800 | 0.08781 |
| tetradecane | 7 | 0.05523 | 0.06845 |
| pentadecane | 8 | 0.05683 | 0.07481 |
| hexadecane | 8 | 0.04841 | 0.06019 |
| heptadecane | 9 | 0.05048 | 0.06515 |
| octadecane | 9 | 0.04310 | 0.05371 |
| nonadecane | 10 | 0.04416 | 0.05769 |
| # C | G | ||||
|---|---|---|---|---|---|
| 3 | propane | 0.82843 | 1.17157 | 0.70711 | 1.00000 |
| cyclopropane | 0.82843 | 1.17157 | |||
| 4 | butane | 0.60815 | 1.00804 | 0.51271 | 0.82878 |
| 2-methylpropane | 0.94183 | 1.49829 | |||
| cyclobutane | 0.59923 | 0.96239 | |||
| methylcyclopropane | 0.92350 | 1.49829 | |||
| 5 | pentane | 0.52109 | 1.00804 | 0.60176 | 1.02855 |
| 2-methylbutane | 0.63713 | 1.24589 | |||
| cyclopentane | 0.97870 | 1.24589 | |||
| methylcyclobutane | 0.56818 | 1.12383 | |||
| 6 | hexane | 0.62511 | 1.01208 | 0.45578 | 0.77120 |
| 2-methylpentane | 1.12838 | 1.84463 | |||
| 3-methylpentane | 0.64722 | 1.10102 | |||
| cyclohexane | 1.08853 | 1.84463 | |||
| methylcyclopentane | 0.71450 | 1.31051 | |||
| 7 | heptane | 0.53898 | 0.93324 | 0.54555 | 0.88169 |
| 2-methylhexane | 0.73555 | 1.26051 | |||
| 3-methylhexane | 0.63149 | 1.10946 | |||
| cycloheptane | 0.86743 | 1.26051 | |||
| methylcyclohexane | 0.66489 | 0.99316 | |||
| 8 | octane | 0.46607 | 0.75497 | 0.49261 | 0.80174 |
| 2-methylheptane | 0.74269 | 1.23178 | |||
| 3-methylheptane | 0.44176 | 0.64669 | |||
| 4-methylheptane | 0.69068 | 1.16592 | |||
| cyclooctane | 0.52925 | 0.89428 | |||
| methylcycloheptane | 0.77031 | 1.23178 | |||
| 9 | nonane | 0.43936 | 0.89004 | 0.46611 | 0.83639 |
| 2-methyloktane | 0.62917 | 1.26588 | |||
| 3-methyloktane | 0.49257 | 1.04540 | |||
| 4-methyloktane | 0.54058 | 1.13356 | |||
| cyclononane | 0.91621 | 1.26588 | |||
| methylcyclooctane | 0.52234 | 0.75185 | |||
| 10 | decane | 0.52739 | 0.89092 | 0.44980 | 0.78752 |
| 2-methylnonane | 0.85978 | 1.60171 | |||
| 3-methylnonane | 0.53326 | 1.00676 | |||
| 4-methylnonane | 0.79253 | 1.51436 | |||
| 5-methylnonane | 0.53671 | 1.02626 | |||
| cyclodecane | 1.06170 | 1.60171 | |||
| methylcyclononane | 0.73172 | 1.18791 | |||
| 11 | undecane | 0.44258 | 0.86184 | 0.47531 | 0.85526 |
| 2-methyldecane | 0.63656 | 1.26844 | |||
| 3-methyldecane | 0.48254 | 1.00747 | |||
| 4-methyldecane | 0.55282 | 1.14338 | |||
| 5-methyldecane | 0.51390 | 1.07445 | |||
| cycloundecane | 0.91944 | 1.26844 | |||
| methylcyclodecane | 0.67251 | 0.96995 |
| # C | G | ||||
|---|---|---|---|---|---|
| 12 | dodecane | 0.43322 | 0.68215 | 0.47928 | 0.79161 |
| 2-methylundecane | 0.65890 | 1.15977 | |||
| 3-methylundecane | 0.40449 | 0.59723 | |||
| 4-methylundecane | 0.58503 | 1.06267 | |||
| 5-methylundecane | 0.40266 | 0.62438 | |||
| 6-methylundecane | 0.56934 | 1.04079 | |||
| cyclododecane | 0.63885 | 1.01789 | |||
| methylcycloundecane | 0.75435 | 1.15977 | |||
| 13 | tridecane | 0.40136 | 0.84197 | 0.42822 | 0.83628 |
| 2-methyldodecane | 0.58248 | 1.26986 | |||
| 3-methyldodecane | 0.42327 | 0.98223 | |||
| 4-methyldodecane | 0.50147 | 1.14839 | |||
| 5-methyldodecane | 0.44474 | 1.03935 | |||
| 6-methyldodecane | 0.46724 | 1.08698 | |||
| cyclotridecane | 0.93790 | 1.26986 | |||
| methylcyclododecane | 0.59177 | 0.85706 | |||
| 14 | tetradecane | 0.47516 | 0.84230 | 0.42032 | 0.79215 |
| 2-methyltridecane | 0.73279 | 1.50436 | |||
| 3-methyltridecane | 0.46608 | 0.96408 | |||
| 4-methyltridecane | 0.65694 | 1.40199 | |||
| 5-methyltridecane | 0.46881 | 0.99504 | |||
| 6-methyltridecane | 0.63480 | 1.37006 | |||
| 7-methyltridecane | 0.46980 | 1.00269 | |||
| cyclotetradecane | 1.04737 | 1.50436 | |||
| methylcyclotridecane | 0.73900 | 1.14013 | |||
| 15 | pentadecane | 0.40023 | 0.82720 | 0.42503 | 0.84107 |
| 2-methyltetradecane | 0.57807 | 1.27073 | |||
| 3-methyltetradecane | 0.41464 | 0.96417 | |||
| 4-methyltetradecane | 0.49861 | 1.1513 | |||
| 5-methyltetradecane | 0.43140 | 1.01587 | |||
| 6-methyltetradecane | 0.46638 | 1.09363 | |||
| 7-methyltetradecane | 0.44703 | 1.05359 | |||
| cyclopentadecane | 0.94172 | 1.27073 | |||
| methylcyclotetradecane | 0.68286 | 0.97177 | |||
| 16 | hexadecane | 0.41172 | 0.64717 | 0.45513 | 0.78507 |
| 2-methylpentadecane | 0.60447 | 1.12561 | |||
| 3-methylpentadecane | 0.37716 | 0.60159 | |||
| 4-methylpentadecane | 0.52563 | 1.02008 | |||
| 5-methylpentadecane | 0.37429 | 0.63474 | |||
| 6-methylpentadecane | 0.50004 | 0.98267 | |||
| 7-methylpentadecane | 0.37398 | 0.64622 | |||
| 8-methylpentadecane | 0.49340 | 0.97257 | |||
| cyclohexadecane | 0.71180 | 1.08063 | |||
| methylcyclopentadecane | 0.75050 | 1.12561 |
| # C | G | ||||
|---|---|---|---|---|---|
| 17 | heptadecane | 0.37776 | 0.81578 | 0.25875 | 0.83038 |
| 2-methylhexadecane | 0.54462 | 1.27131 | |||
| 3-methylhexadecane | 0.38081 | 0.95060 | |||
| 4-methylhexadecane | 0.46564 | 1.15315 | |||
| 5-methylhexadecane | 0.39333 | 0.99899 | |||
| 6-methylhexadecane | 0.43471 | 1.09763 | |||
| 7-methylhexadecane | 0.40470 | 1.03127 | |||
| 8-methylhexadecane | 0.41728 | 1.06138 | |||
| cycloheptadecane | 0.95089 | 1.27131 | |||
| methylcyclohexadecane | 0.63079 | 0.90527 | |||
| 18 | octadecane | 0.44099 | 0.81595 | 0.39023 | 0.79306 |
| 2-methylheptadecane | 0.65567 | 1.45164 | |||
| 3-methylheptadecane | 0.42065 | 0.94002 | |||
| 4-methylheptadecane | 0.57727 | 1.34405 | |||
| 5-methylheptadecane | 0.42136 | 0.97455 | |||
| 6-methylheptadecane | 0.55018 | 1.30334 | |||
| 7-methylheptadecane | 0.42243 | 0.98823 | |||
| 8-methylheptadecane | 0.54024 | 1.28771 | |||
| 9-methylheptadecane | 0.42280 | 0.99206 | |||
| cyclooctadecane | 1.03767 | 1.45164 | |||
| methylcycloheptadecane | 0.74203 | 1.11443 | |||
| 19 | nonadecane | 0.37550 | 0.80672 | 0.38681 | 0.83130 |
| 2-methyloctadecane | 0.53800 | 1.27170 | |||
| 3-methyloctadecane | 0.37328 | 0.94002 | |||
| 4-methyloctadecane | 0.45958 | 1.15440 | |||
| 5-methyloctadecane | 0.38349 | 0.98624 | |||
| 6-methyloctadecane | 0.42938 | 1.10022 | |||
| 7-methyloctadecane | 0.39253 | 1.01519 | |||
| 8-methyloctadecane | 0.41291 | 1.06615 | |||
| 9-methyloctadecane | 0.40171 | 1.03984 | |||
| cyclononadecane | 0.95361 | 1.27170 | |||
| methylcyclooctadecane | 0.69097 | 0.97670 | |||
| 20 | icosane | 0.39502 | 0.62657 | 0.42462 | 0.77814 |
| 2-methylnonadecane | 0.56515 | 1.11860 | |||
| 3-methylnonadecane | 0.35491 | 0.61675 | |||
| 4-methylnonadecane | 0.48431 | 1.00960 | |||
| 5-methylnonadecane | 0.35084 | 0.65221 | |||
| 6-methylnonadecane | 0.45574 | 0.96672 | |||
| 7-methylnonadecane | 0.35019 | 0.66727 | |||
| 8-methylnonadecane | 0.44384 | 0.94775 | |||
| 9-methylnonadecane | 0.35011 | 0.67330 | |||
| 10-methylnonadecane | 0.44046 | 0.94222 | |||
| cycloicosane | 0.76036 | 1.11860 | |||
| methylcyclononadecane | 0.74880 | 1.10555 |
| Set | Molecule | E(G) | Set | Molecule | E(G) | Set | Molecule | E(G) |
|---|---|---|---|---|---|---|---|---|
| ethane | 2.00000 | methylcycloundecane | 15.07008 | 2-methyltetradecane | 17.86281 | |||
| propane | 2.82843 | methylcyclododecane | 16.17966 | 3-methyltetradecane | 18.16937 | |||
| butane | 4.47214 | methylcyclotridecane | 17.61161 | 4-methyltetradecane | 17.98224 | |||
| pentane | 5.46410 | methylcyclotetradecane | 18.83458 | 5-methyltetradecane | 18.11767 | |||
| hexane | 6.98792 | methylcyclopentadecane | 20.15434 | 6-methyltetradecane | 18.03991 | |||
| heptane | 8.05468 | methylcyclohexadecane | 21.30986 | 7-methyltetradecane | 18.07995 | |||
| octane | 9.51754 | methylcycloheptadecane | 22.69787 | 2-methylpentadecane | 19.02873 | |||
| nonane | 10.62750 | methylcyclooctadecane | 23.92413 | 3-methylpentadecane | 19.55275 | |||
| decane | 12.05335 | methylcyclononadecane | 25.24196 | 4-methylpentadecane | 19.13426 | |||
| undecane | 13.19151 | 2-methyilpropane | 3.46410 | 5-methylpentadecane | 19.51960 | |||
| dodecane | 14.59246 | 2-methylbutane | 5.22625 | 6-methylpentadecane | 19.17167 | |||
| tridecane | 15.75049 | 2-methylpentane | 6.15537 | 7-methylpentadecane | 19.50812 | |||
| tetradecane | 17.13354 | 3-methylpentane | 6.89898 | 8-methylpentadecane | 19.18177 | |||
| pentadecane | 18.30634 | 2-methylhexane | 7.72741 | 2-methylhexadecane | 20.40459 | |||
| hexadecane | 19.67590 | 3-methylhexane | 7.87846 | 3-methylhexadecane | 20.72530 | |||
| heptadecane | 20.86010 | 2-methylheptane | 8.76257 | 4-methylhexadecane | 20.52275 | |||
| octadecane | 22.21913 | 3-methylheptane | 9.40926 | 5-methylhexadecane | 20.67691 | |||
| nonadecane | 23.41241 | 4-methylheptane | 8.82843 | 6-methylhexadecane | 20.57827 | |||
| icosane | 24.76298 | 2-methyloktane | 10.25166 | 7-methylhexadecane | 20.64463 | |||
| cyclopropane | 4.00000 | 3-methyloktane | 10.47214 | 8-methylhexadecane | 20.61452 | |||
| cyclobutane | 4.00000 | 4-methyloktane | 10.38398 | 2-methylheptadecane | 21.58344 | |||
| cyclopentane | 6.47214 | 2-methylnonane | 11.34256 | 3-methylheptadecane | 22.09506 | |||
| cyclohexane | 8.00000 | 3-methylnonane | 11.93751 | 4-methylheptadecane | 21.69103 | |||
| cycloheptane | 8.98792 | 4-methylnonane | 11.42991 | 5-methylheptadecane | 22.06053 | |||
| cyclooctane | 9.65685 | 5-methylnonane | 11.91801 | 6-methylheptadecane | 21.73174 | |||
| cyclononane | 11.51754 | 2-methyldecane | 12.78491 | 7-methylheptadecane | 22.04685 | |||
| cyclodecane | 12.94427 | 3-methyldecane | 13.04588 | 8-methylheptadecane | 21.74737 | |||
| cycloundecane | 14.05335 | 4-methyldecane | 12.90997 | 9-methylheptadecane | 22.04302 | |||
| cyclododecane | 14.92820 | 5-methyldecane | 12.97890 | 2-methyloctadecane | 22.94743 | |||
| cyclotridecane | 16.59246 | 2-methylundecane | 13.91031 | 3-methyloctadecane | 23.27911 | |||
| cyclotetradecane | 17.97584 | 3-methylundecane | 14.47285 | 4-methyloctadecane | 23.06473 | |||
| cyclopentadecane | 19.13354 | 4-methylundecane | 14.00741 | 5-methyloctadecane | 23.23289 | |||
| cyclohexadecane | 20.10936 | 5-methylundecane | 14.44570 | 6-methyloctadecane | 23.11891 | |||
| cycloheptadecane | 21.67590 | 6-methylundecane | 14.02929 | 7-methyloctadecane | 23.20394 | |||
| cyclooctadecane | 23.03508 | 2-methyldodecane | 15.32260 | 8-methyloctadecane | 23.15298 | |||
| cyclononadecane | 24.21913 | 3-methyldodecane | 15.61023 | 9-methyloctadecane | 23.17929 | |||
| cycloicosane | 25.25501 | 4-methyldodecane | 15.44407 | 2-methylnonadecane | 24.13641 | |||
| methylcyclopropane | 4.96239 | 5-methyldodecane | 15.55311 | 3-methylnonadecane | 24.63826 | |||
| methylcyclobutane | 5.34831 | 6-methyldodecane | 15.50548 | 4-methylnonadecane | 24.24541 | |||
| methylcyclopentane | 7.46588 | 2-methyltridecane | 16.47148 | 5-methylnonadecane | 24.60280 | |||
| methylcyclohexane | 8.72057 | 3-methyltridecane | 17.01176 | 6-methylnonadecane | 24.28829 | |||
| methylcycloheptane | 9.99435 | 4-methyltridecane | 16.57385 | 7-methylnonadecane | 24.58774 | |||
| methylcyclooctane | 11.00351 | 5-methyltridecane | 16.98080 | 8-methylnonadecane | 24.30726 | |||
| methylcyclononane | 12.53047 | 6-methyltridecane | 16.60578 | 9-methylnonadecane | 24.58171 | |||
| methylcyclodecane | 13.75486 | 7-methyltridecane | 16.97315 | 10-methylnonadecane | 24.31279 |
References
- Gutman, I. The energy of a graph. Ber. Math-Statist. Sekt. Forchungsz. Graz 1978, 103, 1–22. [Google Scholar]
- Gutman, I. The energy of a Graph: Old and New Results. Algebraic Combinatorics and Applications; Springer: Berlin/Heidelberg, Germany, 2001; pp. 196–211. [Google Scholar]
- Li, X.; Shi, Y.; Gutman, I. Graph Energy; Springer Science & Business Mediam: New York, NY, USA, 2012. [Google Scholar]
- Gutman, I.; Li, X.; Zhang, J. Graph energy. In Analysis of Complex Networks: From Biology to Linguistics; Wiley-VCH: Weinheim, Germany, 2009; pp. 145–174. [Google Scholar]
- Hückel, E. Quantentheoretische Beiträge zum Benzolproblem. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Coulson, C.A.; O‘Leary, B.; Mallion, R.B. Hückel Theory for Organic Chemists; Academic Press: London, UK, 1987. [Google Scholar]
- Coulson, C.A. On the calculation of the energy in unsaturated hydrocarbon molecules. Proc. Camb. Phil. Soc. 1940, 36, 201–203. [Google Scholar] [CrossRef]
- McCelland, B.J. Properties of the latent roots of a matrix: The estimation of π-electron energies. J. Chem. Phys. 1971, 54, 640–643. [Google Scholar] [CrossRef]
- Koolen, J.H.; Moulton, V.; Gutman, I. Improving the McClelland inequality for total π-electron energy. Chem. Phys. Lett. 2000, 320, 213–216. [Google Scholar] [CrossRef]
- Estrada, E.; Benzi, M. What is the meaning of the graph energy after all? Discrete Appl. Math. 2017, 230, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Gutman, I.; Furtula, B. Graph energies and their applications. Bull. Sci. Math. 2019, 44, 29–45. [Google Scholar]
- Furtula, B.; Gutman, I.; Dehmer, M. On structure-sensitivity ofdegree-based topological indices. Appl. Math. Comput. 2013, 219, 8973–8978. [Google Scholar]
- Gao, X.; Xiao, B.; Tao, D. A survey of graph edit distance. Pattern Anal. Appl. 2010, 13, 113–129. [Google Scholar] [CrossRef]
- Rakić, M.; Furtula, B. A novel method for measuring the structure sensitivity of molecular descriptors. J. Chemom. 2019, 33, e3138. [Google Scholar] [CrossRef]
- Redžepović, I.; Furtula, B. Comparative study on structural sensitivity of eigenvalue–based molecular descriptors. J. Math. Chem. 2021, 59, 476–487. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zemljič, K.; Žigert Pleteršek, P. Smoothness of Graph Energy in Chemical Graphs. Mathematics 2023, 11, 552. https://doi.org/10.3390/math11030552
Zemljič K, Žigert Pleteršek P. Smoothness of Graph Energy in Chemical Graphs. Mathematics. 2023; 11(3):552. https://doi.org/10.3390/math11030552
Chicago/Turabian StyleZemljič, Katja, and Petra Žigert Pleteršek. 2023. "Smoothness of Graph Energy in Chemical Graphs" Mathematics 11, no. 3: 552. https://doi.org/10.3390/math11030552
APA StyleZemljič, K., & Žigert Pleteršek, P. (2023). Smoothness of Graph Energy in Chemical Graphs. Mathematics, 11(3), 552. https://doi.org/10.3390/math11030552