

Vaginal and Uterine Microbiota of Healthy Maiden Mares during Estrus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Farm Management and Mares
2.3. Sample Collection
2.4. DNA Extraction and V3–V4 Sequencing
2.5. Data Analysis
3. Results
3.1. Overall Sequence Analysis
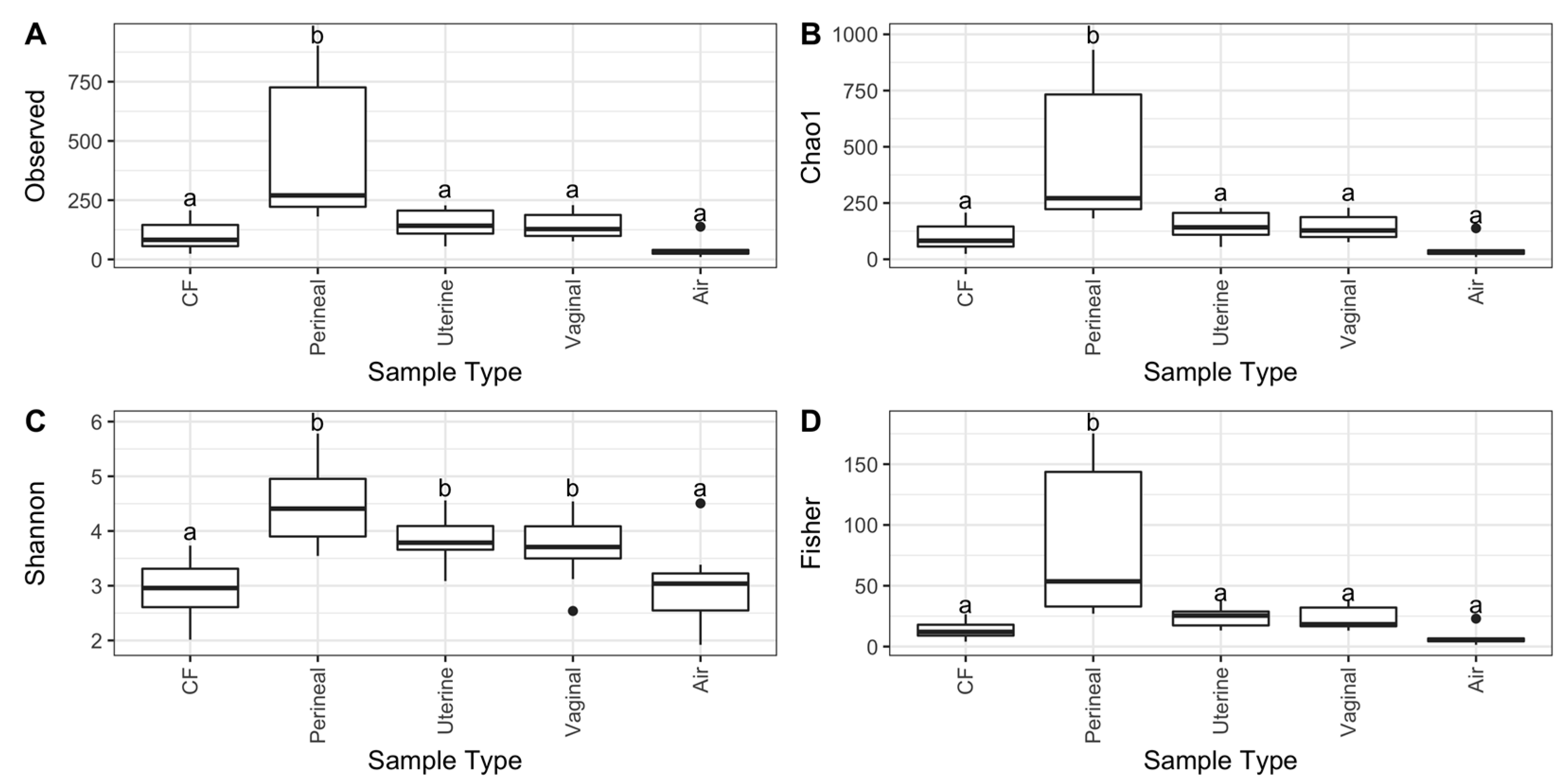
3.2. Microbial Diversity of the Equine Reproductive Tract
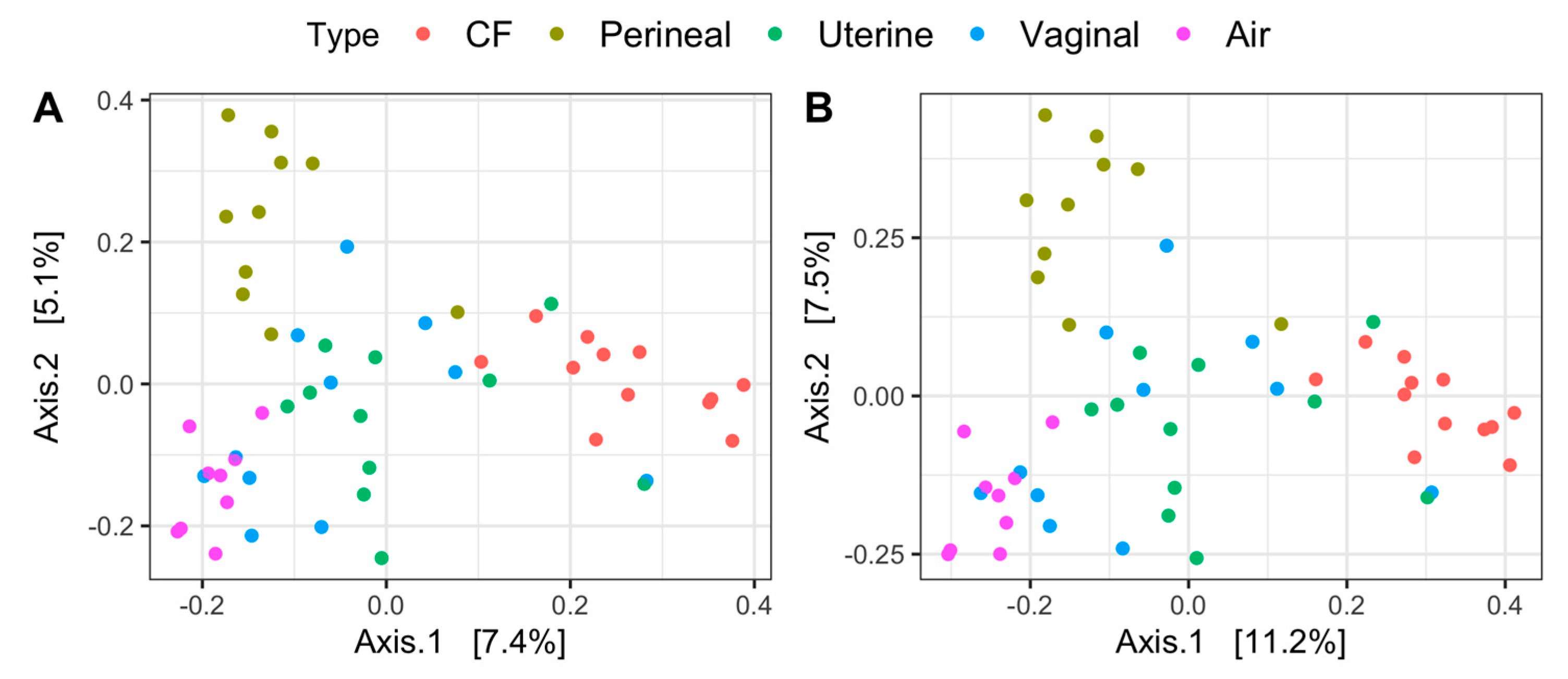
3.3. B-Diversity Analysis
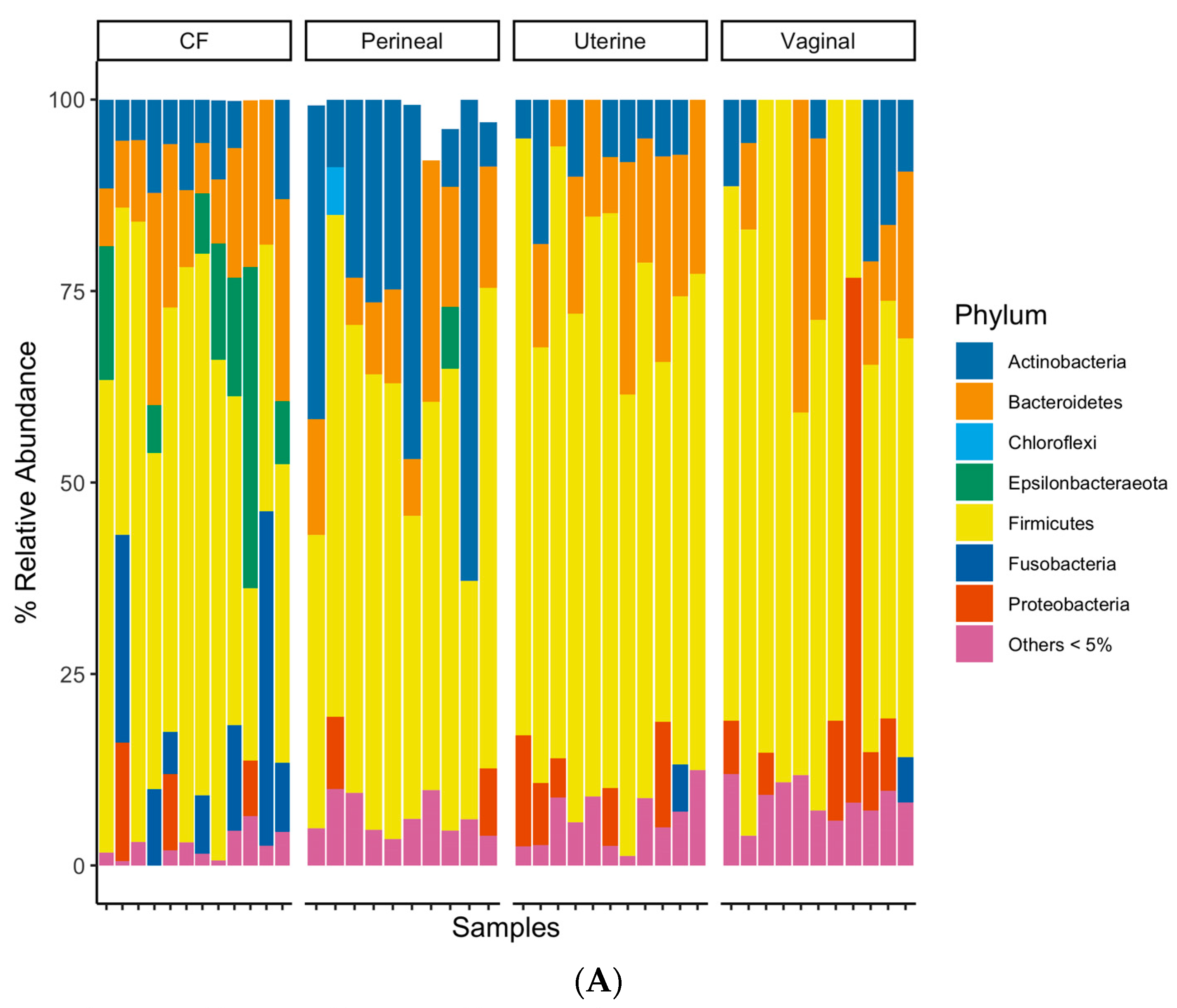
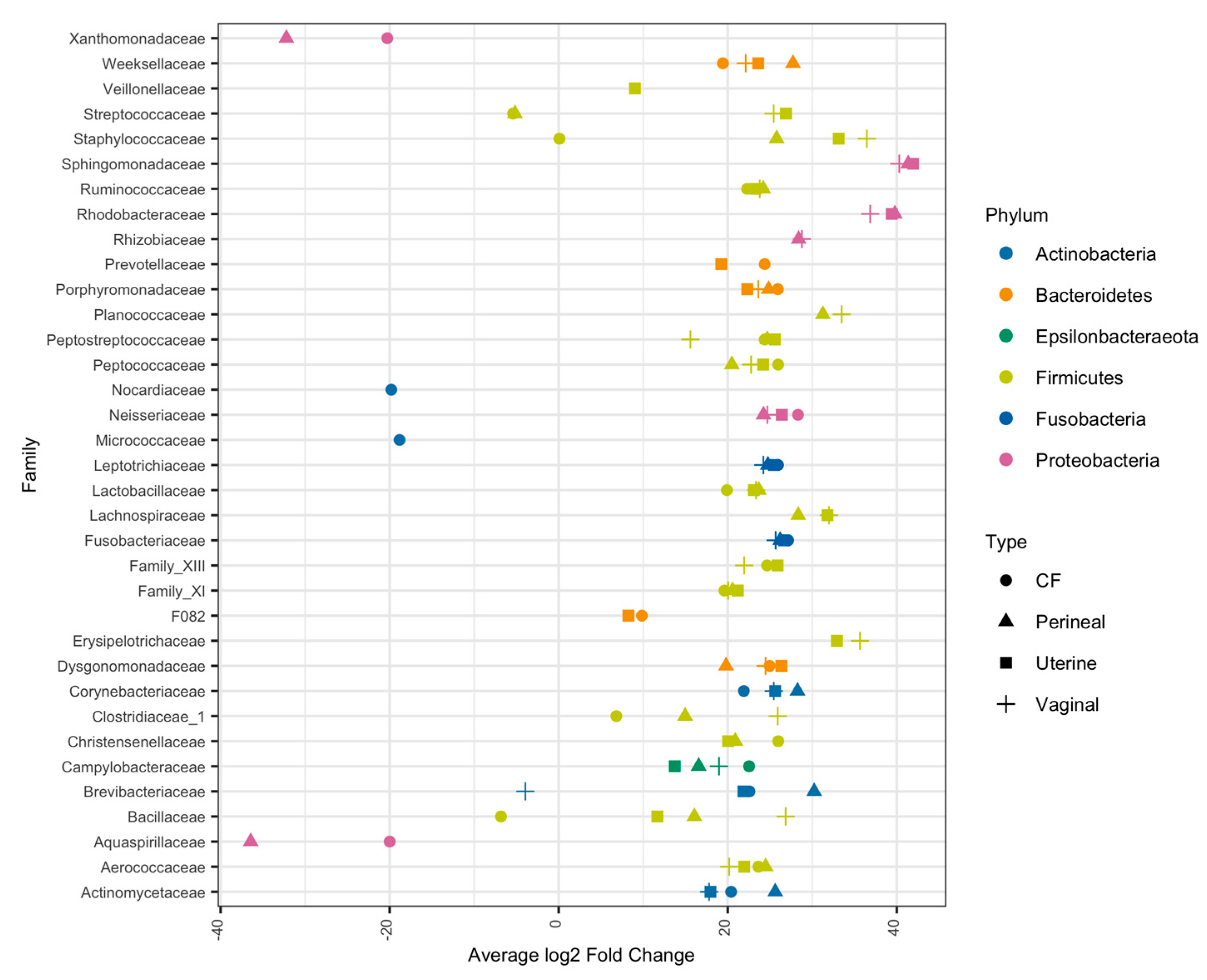
3.4. Taxonomic Composition of Equine Reproductive Anatomy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banchi, P.; Spanoghe, L.; Maes, D.; Morrell, J.; Van Soom, A. The Reproductive Microbiome in Dogs: Friend or Foe? Vet. J. 2024, 304, 106100. [Google Scholar] [CrossRef] [PubMed]
- Al-Nasiry, S.; Ambrosino, E.; Schlaepfer, M.; Morré, S.A.; Wieten, L.; Voncken, J.W.; Spinelli, M.; Mueller, M.; Kramer, B.W. The Interplay between Reproductive Tract Microbiota and Immunological System in Human Reproduction. Front. Immunol. 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Galvão, K.N.; Rodriguez-Lecompte, J.C.; Costa, M.C. The Cattle Microbiota and the Immune System: An Evolving Field. Vet. Clin. N. Am. Food Anim. Pract. 2019, 35, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.; Simon, C. Deciphering the Effect of Reproductive Tract Microbiota on Human Reproduction. Reprod. Med. Biol. 2019, 18, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, B.A.; Reese, S.T.; Dantas, F.G.; Franco, G.A.; Smith, T.P.L.; Adeyosoye, O.I.; Pohler, K.G.; Myer, P.R. Vaginal and Uterine Bacterial Communities in Postpartum Lactating Cows. Front. Microbiol. 2017, 8, 1047. [Google Scholar] [CrossRef] [PubMed]
- Molina, N.M.; Sola-Leyva, A.; Haahr, T.; Aghajanova, L.; Laudanski, P.; Castilla, J.A.; Altmäe, S. Analysing Endometrial Microbiome: Methodological Considerations and Recommendations for Good Practice. Hum. Reprod. 2021, 36, 859–879. [Google Scholar] [CrossRef] [PubMed]
- Molina, N.M.; Sola-Leyva, A.; Saez-Lara, M.J.; Plaza-Diaz, J.; Tubić-Pavlović, A.; Romero, B.; Clavero, A.; Mozas-Moreno, J.; Fontes, J.; Altmäe, S. New Opportunities for Endometrial Health by Modifying Uterine Microbial Composition: Present or Future? Biomolecules 2020, 10, 593. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The Microbiota Continuum along the Female Reproductive Tract and Its Relation to Uterine-Related Diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.M.A.; Gilbert, R.O.; Bicalho, R.C. Metagenomic Analysis of the Uterine Bacterial Microbiota in Healthy and Metritic Postpartum Dairy Cows. J. Dairy Sci. 2011, 94, 291–302. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Y.; Mi, J.; Zhao, Y.; Holyoak, G.R.; Yi, Z.; Wu, R.; Wang, Z.; Zeng, S. Endometrial and Vaginal Microbiome in Donkeys with and without Clinical Endometritis. Front. Microbiol. 2022, 13, 884574. [Google Scholar] [CrossRef]
- LeBlanc, M.M.; Causey, R.C. Clinical and Subclinical Endometritis in the Mare: Both Threats to Fertility. Reprod. Domest. Anim. 2009, 44 (Suppl. S3), 10–22. [Google Scholar] [CrossRef] [PubMed]
- Barba, M.; Martínez-Boví, R.; Quereda, J.J.; Mocé, M.L.; Plaza-Dávila, M.; Jiménez-Trigos, E.; Gómez-Martín, Á.; González-Torres, P.; Carbonetto, B.; García-Roselló, E.; et al. Comparison of Two Techniques for Obtaining Endometrial Bacteriologic Cultures in the Mare. Theriogenology 2022, 10, 85–93. [Google Scholar] [CrossRef]
- Beckers, K.F.; Gomes, V.C.L.; Crissman, K.R.; Liu, C.C.; Schulz, C.J.; Childers, G.W.; Sones, J.L. Metagenetic Analysis of the Pregnant Microbiome in Horses. Animals 2023, 13, 1999. [Google Scholar] [CrossRef]
- Heil, B.A.; van Heule, M.; Thompson, S.K.; Kearns, T.A.; Oberhaus, E.L.; King, G.; Daels, P.; Dini, P.; Sones, J.L. Effect of Sampling Method on Detection of the Equine Uterine Microbiome during Estrus. Vet. Sci. 2023, 10, 644. [Google Scholar] [CrossRef]
- Holyoak, G.R.; Premathilake, H.U.; Lyman, C.C.; Sones, J.L.; Gunn, A.; Wieneke, X.; DeSilva, U. The Healthy Equine Uterus Harbors a Distinct Core Microbiome plus a Rich and Diverse Microbiome That Varies with Geographical Location. Sci. Rep. 2022, 12, 14790. [Google Scholar] [CrossRef]
- Poole, R.K.; Soffa, D.R.; McAnally, B.E.; Smith, M.S.; Hickman-Brown, K.J.; Stockland, E.L. Reproductive Microbiomes in Domestic Livestock: Insights Utilizing 16S RRNA Gene Amplicon Community Sequencing. Animals 2023, 13, 485. [Google Scholar] [CrossRef]
- Webb, E.M.; Holman, D.B.; Schmidt, K.N.; Pun, B.; Sedivec, K.K.; Hurlbert, J.L.; Bochantin, K.A.; Ward, A.K.; Dahlen, C.R.; Amat, S. Sequencing and Culture-Based Characterization of the Vaginal and Uterine Microbiota in Beef Cattle That Became Pregnant or Remained Open Following Artificial Insemination. Microbiol. Spectr. 2023, 11, e0273223. [Google Scholar] [CrossRef] [PubMed]
- Heil, B.A.; van Heule, M.; Thompson, S.; Kearns, T.A.; Beckers Kalie, F.; Oberhaus, L.; King, G.; Daels, P.; Dini, P.; Sones, J.L. Metagenomic Characterization of the Equine Endometrial Microbiome During Anestrus. J. Equine Vet. Sci. 2024, 140, 105134. [Google Scholar] [CrossRef]
- Zakia, L.S.; Gomez, D.E.; Caddey, B.B.; Boerlin, P.; Surette, M.G.; Arroyo, L.G. Direct and Culture-Enriched 16S RRNA Sequencing of Cecal Content of Healthy Horses and Horses with Typhlocolitis. PLoS ONE 2023, 18, e0284193. [Google Scholar] [CrossRef]
- Bartram, A.K.; Lynch, M.D.J.; Stearns, J.C.; Moreno-Hagelsieb, G.; Neufeld, J.D. Generation of Multimillion-Sequence 16S RRNA Gene Libraries from Complex Microbial Communities by Assembling Paired-End Illumina Reads. Appl. Environ. Microbiol. 2011, 77, 3846–3852. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian Classifier for Rapid Assignment of RRNA Sequences into the New Bacterial Taxonomy. Appl. Env. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic. Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Silva, J.A.; Castañares, M.; Mouguelar, H.; Valenciano, J.A.; Pellegrino, M.S. Isolation of Lactic Acid Bacteria from the Reproductive Tract of Mares as Potentially Beneficial Strains to Prevent Equine Endometritis. Vet. Res. Commun. 2024, 48, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Kudo, H.; Cheng, K.J.; Costerton, J.W. Interactions between Treponema Bryantii and Cellulolytic Bacteria in the in Vitro Degradation of Straw Cellulose. Can. J. Microbiol. 1987, 33, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Scarlet, D.; Malama, E.; Fischer, S.; Knutti, B.; Bollwein, H. Relationship between Clinical Uterine Findings, Therapy, and Fertility in the Mare. Vet. Sci. 2023, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Turni, C.; Blackall, P.J.; Boe-Hansen, G.; Hayes, B.J.; Tabor, A.E. Interrogating the Bovine Reproductive Tract Metagenomes Using Culture-Independent Approaches: A Systematic Review. Anim. Microbiome 2021, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Thomson, P.; Pareja, J.; Núñez, A.; Santibáñez, R.; Castro, R. Characterization of Microbial Communities and Predicted Metabolic Pathways in the Uterus of Healthy Mares. Open Vet. J. 2022, 12, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Weese, J.S. Understanding the Intestinal Microbiome in Health and Disease. Vet. Clin. N. Am. Equine Pract. 2018, 34, 1–12. [Google Scholar] [CrossRef]
- Gomez, D.E.; Arroyo, L.G.; Lillie, B.; Weese, J.S. Nasal Bacterial Microbiota during an Outbreak of Equine Herpesvirus 1 at a Farm in Southern Ontario. Can J. Vet. Res. 2021, 85, 3–11. [Google Scholar] [PubMed]
- Dahl, W.J.; Rivero Mendoza, D.; Lambert, J.M. Diet, Nutrients and the Microbiome. Prog. Mol. Biol. Transl. Sci. 2020, 171, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Sirichoat, A.; Sankuntaw, N.; Engchanil, C.; Buppasiri, P.; Faksri, K.; Namwat, W.; Chantratita, W.; Lulitanond, V. Comparison of Different Hypervariable Regions of 16S RRNA for Taxonomic Profiling of Vaginal Microbiota Using Next-Generation Sequencing. Arch. Microbiol. 2021, 203, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Mattei, V.; Murugesan, S.; Al Hashmi, M.; Mathew, R.; James, N.; Singh, P.; Kumar, M.; Lakshmanan, A.P.; Terranegra, A.; Al Khodor, S.; et al. Evaluation of Methods for the Extraction of Microbial DNA From Vaginal Swabs Used for Microbiome Studies. Front. Cell Infect. Microbiol. 2019, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-P.; Chen, W.-C.; Cheng, C.-M.; Shen, C.-J.; Vaginal, P.H. Value for Clinical Diagnosis and Treatment of Common Vaginitis. Diagnostics 2021, 11, 1996. [Google Scholar] [CrossRef] [PubMed]
- Barb, J.J.; Oler, A.J.; Kim, H.-S.; Chalmers, N.; Wallen, G.R.; Cashion, A.; Munson, P.J.; Ames, N.J. Development of an Analysis Pipeline Characterizing Multiple Hypervariable Regions of 16S RRNA Using Mock Samples. PLoS ONE 2016, 11, e0148047. [Google Scholar] [CrossRef]
- Miller, E.A.; Livermore, J.A.; Alberts, S.C.; Tung, J.; Archie, E.A. Ovarian Cycling and Reproductive State Shape the Vaginal Microbiota in Wild Baboons. Microbiome 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.G.; Ericsson, A.C.; Poock, S.E.; Melendez, P.; Lucy, M.C. Hot Topic: 16S RRNA Gene Sequencing Reveals the Microbiome of the Virgin and Pregnant Bovine Uterus. J. Dairy Sci. 2017, 100, 4953–4960. [Google Scholar] [CrossRef]
- Lorenzen, E.; Kudirkiene, E.; Gutman, N.; Grossi, A.B.; Agerholm, J.S.; Erneholm, K.; Skytte, C.; Dalgaard, M.D.; Bojesen, A.M. The Vaginal Microbiome Is Stable in Prepubertal and Sexually Mature Ellegaard Göttingen Minipigs throughout an Estrous Cycle. Vet. Res. 2015, 46, 125. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, C.; Arroyo, L.G.; MacNicol, J.L.; Renaud, D.; Weese, J.S.; Gomez, D.E. Fecal Microbiota of Horses with Colitis and Its Association with Laminitis and Survival during Hospitalization. J. Vet. Intern Med. 2022, 36, 2213–2223. [Google Scholar] [CrossRef]
- Gomez, D.E.; Wong, D.; MacNicol, J.; Dembek, K. The Fecal Bacterial Microbiota of Healthy and Sick Newborn Foals. J. Vet. Intern. Med. 2023, 37, 315–322. [Google Scholar] [CrossRef]
- Martin de Bustamante, M.; Gomez, D.; MacNicol, J.; Hamor, R.; Plummer, C. The Fecal Bacterial Microbiota in Horses with Equine Recurrent Uveitis. Animals 2021, 11, 745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, L.; He, Y.; Luo, X.; Zhao, S.; Jia, X. Composition of Fecal Microbiota in Grazing and Feedlot Angus Beef Cattle. Animals 2021, 11, 3167. [Google Scholar] [CrossRef]
- Laguardia-Nascimento, M.; Branco, K.M.G.R.; Gasparini, M.R.; Giannattasio-Ferraz, S.; Leite, L.R.; Araujo, F.M.G.; de Matos Salim, A.C.; Nicoli, J.R.; de Oliveira, G.C.; Barbosa-Stancioli, E.F. Vaginal Microbiome Characterization of Nellore Cattle Using Metagenomic Analysis. PLoS ONE 2015, 10, e0143294. [Google Scholar] [CrossRef]
- Takada, K.; Melnikov, V.G.; Kobayashi, R.; Komine-Aizawa, S.; Tsuji, N.M.; Hayakawa, S. Female Reproductive Tract-Organ Axes. Front. Immunol. 2023, 14, 1110001. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.; Franasiak, J.M. Endometrial Microbiota-New Player in Town. Fertil. Steril. 2017, 108, 32–39. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.; Nesengani, L.T.; Gong, Y.; Zhang, S.; Lu, W. Characterization of Vaginal Microbiota of Endometritis and Healthy Sows Using High-Throughput Pyrosequencing of 16S RRNA Gene. Microb. Pathog. 2017, 111, 325–330. [Google Scholar] [CrossRef]
- Song, Y.G.; Guevarra, R.B.; Lee, J.H.; Wattanaphansak, S.; Kang, B.N.; Kim, H.B.; Song, K.H. Comparative Analysis of the Reproductive Tract Microbial Communities in Female Dogs with and without Pyometra through the 16S RRNA Gene Pyrosequencing. Jpn. J. Vet. Res. 2017, 65, 193–200. [Google Scholar] [CrossRef]
- Lyman, C.C.; Holyoak, G.R.; Meinkoth, K.; Wieneke, X.; Chillemi, K.A.; DeSilva, U. Canine Endometrial and Vaginal Microbiomes Reveal Distinct and Complex Ecosystems. PLoS ONE 2019, 14, e0210157. [Google Scholar] [CrossRef]
- Hinrichs, K.; Cummings, M.R.; Sertich, P.L.; Kenney, R.M. Clinical Significance of Aerobic Bacterial Flora of the Uterus, Vagina, Vestibule, and Clitoral Fossa of Clinically Normal Mares. J. Am. Vet. Med. Assoc. 1988, 193, 72–75. [Google Scholar]
- Virendra, A.; Gulavane, S.U.; Ahmed, Z.A.; Reddy, R.; Chaudhari, R.J.; Gaikwad, S.M.; Shelar, R.R.; Ingole, S.D.; Thorat, V.D.; Khanam, A.; et al. Metagenomic analysis unravels novel taxonomic differences in the uterine microbiome between healthy mares and mares with endometritis. Vet. Med. Sci. 2024, 10, e1369. [Google Scholar] [CrossRef]
- LeBlanc, S.J. Review: Postpartum Reproductive Disease and Fertility in Dairy Cows. Animal 2023, 17 (Suppl. S1), 100781. [Google Scholar] [CrossRef]
- Canisso, I.F.; Segabinazzi, L.G.T.M.; Fedorka, C.E. Persistent Breeding-Induced Endometritis in Mares—A Multifaceted Challenge: From Clinical Aspects to Immunopathogenesis and Pathobiology. Int. J. Mol. Sci. 2020, 21, 1432. [Google Scholar] [CrossRef]
- Timoney, J.F. The Pathogenic Equine Streptococci. Vet. Res. 2004, 35, 397–409. [Google Scholar] [CrossRef]
- H A Morris, L.; M McCue, P.; Aurich, C. Equine Endometritis: A Review of Challenges and New Approaches. Reproduction 2020, 160, R95–R110. [Google Scholar] [CrossRef]
- Freitas, A.C.; Chaban, B.; Bocking, A.; Rocco, M.; Yang, S.; Hill, J.E.; Money, D.M. The Vaginal Microbiome of Pregnant Women Is Less Rich and Diverse, with Lower Prevalence of Mollicutes, Compared to Non-Pregnant Women. Sci. Rep. 2017, 7, 9212. [Google Scholar] [CrossRef]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.-A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A.; et al. A Metagenomic Approach to Characterization of the Vaginal Microbiome Signature in Pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Gholiof, M.; Adamson-De Luca, E.; Wessels, J.M. The Female Reproductive Tract Microbiotas, Inflammation, and Gynecological Conditions. Front. Reprod. Health 2022, 4, 963752. [Google Scholar] [CrossRef]
- Punzón-Jiménez, P.; Labarta, E. The Impact of the Female Genital Tract Microbiome in Women Health and Reproduction: A Review. J. Assist. Reprod. Genet. 2021, 38, 2519–2541. [Google Scholar] [CrossRef]
- Lehtoranta, L.; Ala-Jaakkola, R.; Laitila, A.; Maukonen, J. Healthy Vaginal Microbiota and Influence of Probiotics Across the Female Life Span. Front. Microbiol. 2022, 13, 819958. [Google Scholar] [CrossRef]
- Amabebe, E.; Anumba, D.O.C. The Vaginal Microenvironment: The Physiologic Role of Lactobacilli. Front. Med. 2018, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Scott, R.T.J. Reproductive Tract Microbiome in Assisted Reproductive Technologies. Fertil. Steril. 2015, 104, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Stoyancheva, G.; Marzotto, M.; Dellaglio, F.; Torriani, S. Bacteriocin Production and Gene Sequencing Analysis from Vaginal Lactobacillus Strains. Arch. Microbiol. 2014, 196, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Malaluang, P.; Åkerholm, T.; Nyman, G.; Lindahl, J.; Hansson, I.; Morrell, J.M. Bacteria in the Healthy Equine Vagina during the Estrous Cycle. Theriogenology 2024, 213, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Malaluang, P.; Wilén, E.; Frosth, S.; Lindahl, J.; Hansson, I.; Morrell, J.M. Vaginal Bacteria in Mares and the Occurrence of Antimicrobial Resistance. Microorganisms 2022, 10, 2204. [Google Scholar] [CrossRef]
- Evans, M.J.; Hamer, J.M.; Gason, L.M.; Graham, C.S.; Asbury, A.C.; Irvine, C.H. Clearance of Bacteria and Non-Antigenic Markers Following Intra-Uterine Inoculation into Maiden Mares: Effect of Steroid Hormone Environment. Theriogenology 1986, 26, 37–50. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil-Miranda, A.; Caddey, B.; Orellana-Guerrero, D.; Smith, H.; Samper, J.C.; Gomez, D.E. Vaginal and Uterine Microbiota of Healthy Maiden Mares during Estrus. Vet. Sci. 2024, 11, 323. https://doi.org/10.3390/vetsci11070323
Gil-Miranda A, Caddey B, Orellana-Guerrero D, Smith H, Samper JC, Gomez DE. Vaginal and Uterine Microbiota of Healthy Maiden Mares during Estrus. Veterinary Sciences. 2024; 11(7):323. https://doi.org/10.3390/vetsci11070323
Chicago/Turabian StyleGil-Miranda, Ana, Benjamin Caddey, Daniela Orellana-Guerrero, Hanna Smith, Juan C. Samper, and Diego E. Gomez. 2024. "Vaginal and Uterine Microbiota of Healthy Maiden Mares during Estrus" Veterinary Sciences 11, no. 7: 323. https://doi.org/10.3390/vetsci11070323
APA StyleGil-Miranda, A., Caddey, B., Orellana-Guerrero, D., Smith, H., Samper, J. C., & Gomez, D. E. (2024). Vaginal and Uterine Microbiota of Healthy Maiden Mares during Estrus. Veterinary Sciences, 11(7), 323. https://doi.org/10.3390/vetsci11070323