
Deletion of the Class 1 Histone Deacetylase PsHos2 Induces Secondary Metabolic Perturbations in the Sea Cucumber-Associated Penicillium sclerotiorum

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. Bioinformatics Analysis
2.3. Plasmids Construction
2.4. P. sclerotiorum SD-36 Transformation
2.5. SM Analysis of P. sclerotiorum SD-36
2.6. Molecular Networking Analysis
2.7. Transcriptome Analysis
2.8. Real-Time PCR Analysis
3. Results
3.1. Gene Sequencing and Bioinformation Analysis
3.2. Generation of the HDAC Gene Knockout Mutant Strain
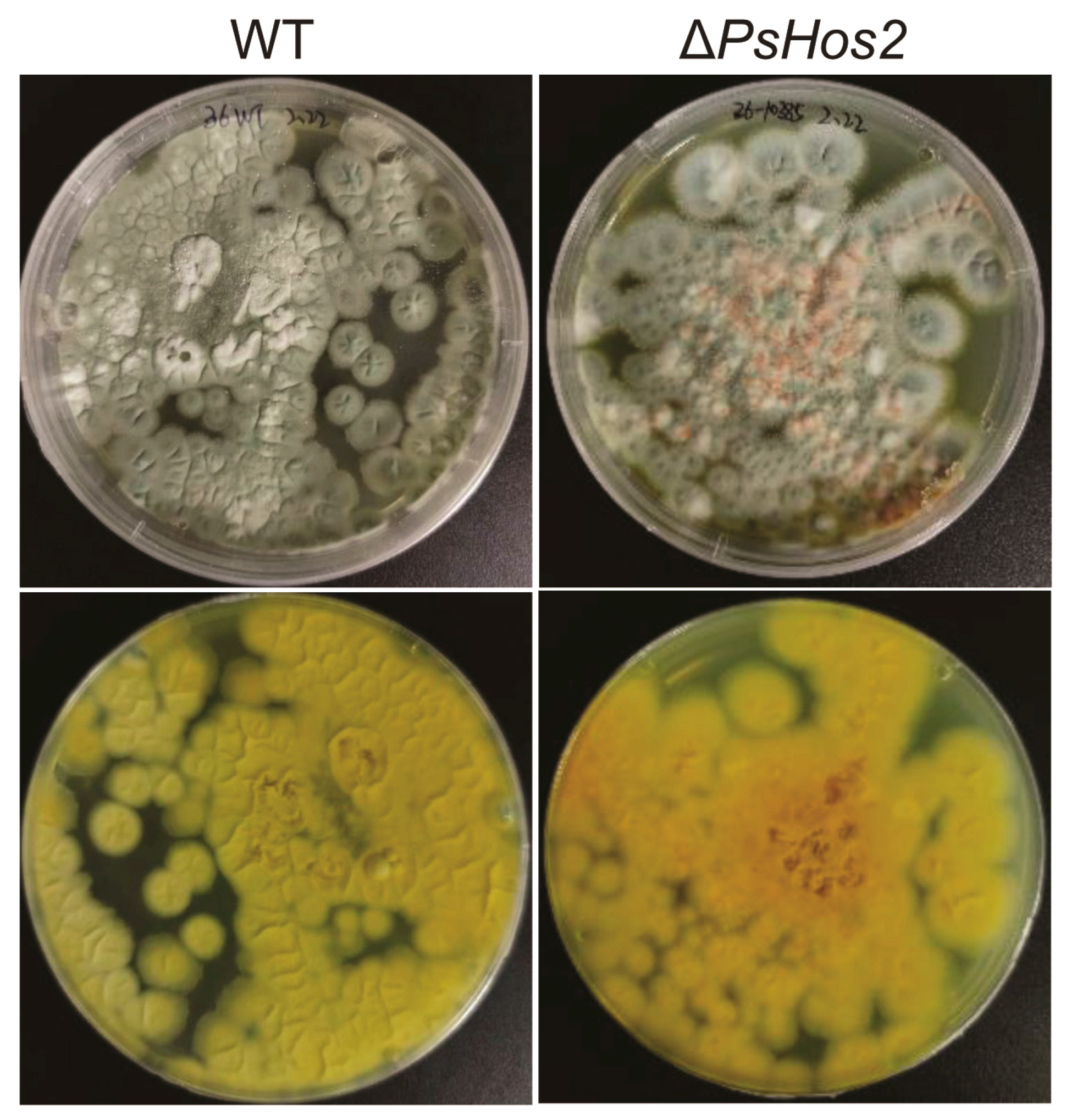
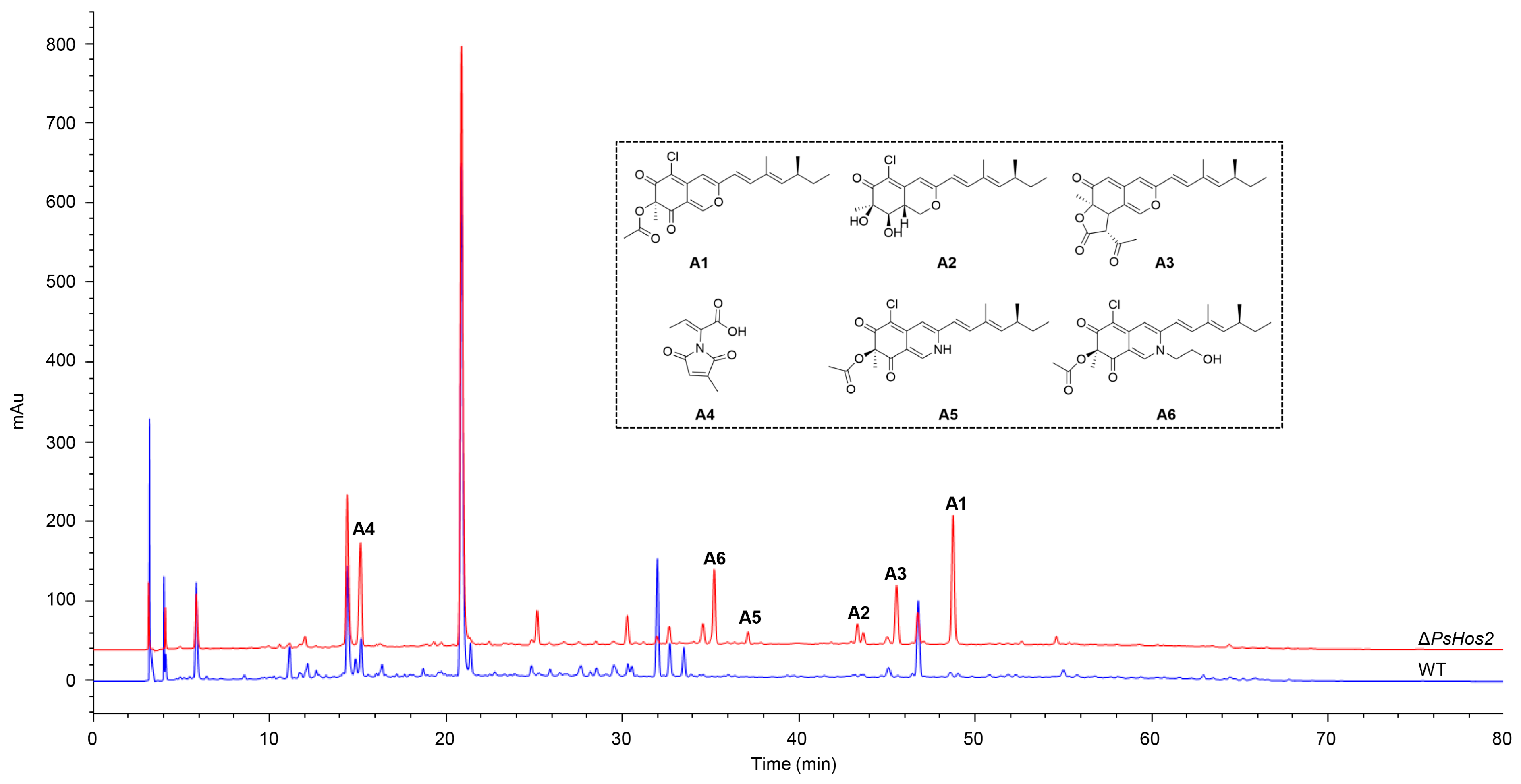
3.3. Effect of HDAC Deletion on the Phenotype, Growth, and SMs of P. sclerotiorum SD-36
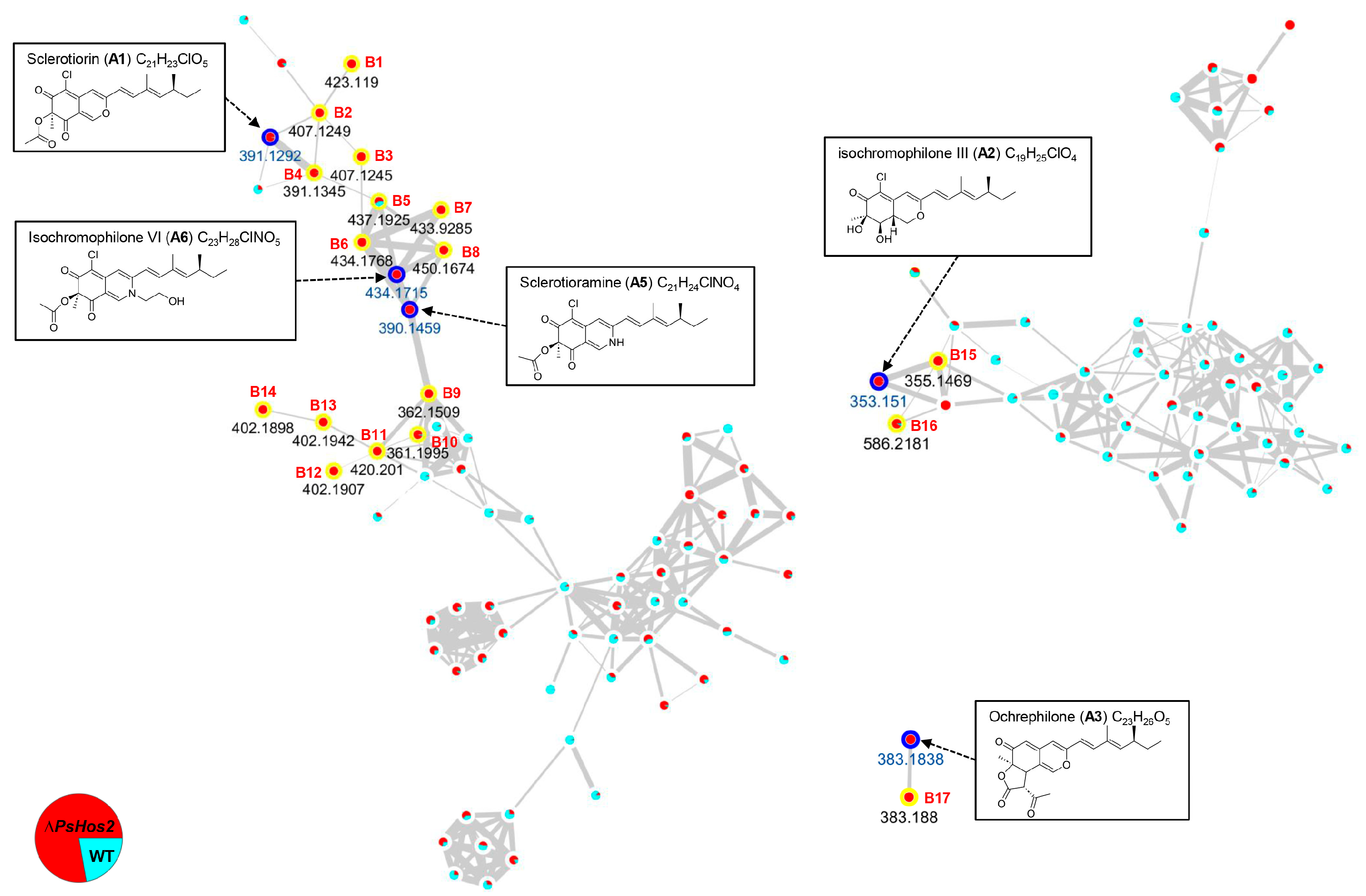
3.4. 17 Potential Azaphilone-Related Nodes Identified by Molecular Networking Analysis
3.5. Putative BGC of Azaphilones in P. sclerotiorum SD-36
3.6. Effect of PsHos2 on the Transcriptome of P. sclerotiorum SD-36
3.7. Real-Time PCR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BGC | Biosynthetic gene cluster |
| HDAC | Histone deacetylase |
| PKS | Polyketide synthase |
| SM | Secondary metabolite |
| PDA | Potato dextrose agar |
| LB | Luria–Bertani medium |
| SSN | Sequence similarity network |
| EtOAc | Ethyl acetate |
| MeOH | Methanol |
| HPLC | High-performance liquid chromatography |
| MeCN | Acetonitrile |
| DCM | Dichloromethane |
| NMR | Nuclear Magnetic Resonance |
| HR-ESI-MS | High-Resolution Electrospray Ionization Mass Spectrometry |
| FBMN | Feature-based molecular networking |
| NRPS | Non-ribosomal peptide synthetase |
| NR-PKS | Non-reducing polyketide synthase |
| HR-PKS | Highly reducing polyketide synthase |
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Meena, S.N.; Wajs-Bonikowska, A.; Girawale, S.; Imran, M.; Poduval, P.; Kodam, K.M. High-throughput mining of novel compounds from known microbes: A boost to natural product screening. Molecules 2024, 29, 3237. [Google Scholar] [CrossRef]
- Willems, T.; De Mol, M.L.; De Bruycker, A.; De Maeseneire, S.L.; Soetaert, W.K. Alkaloids from marine fungi: Promising antimicrobials. Antibiotics 2020, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, J.; Wang, P.; Ding, W. Novel indole diketopiperazine stereoisomers from a marine-derived fungus Aspergillus sp. Mycology 2023, 14, 1–10. [Google Scholar] [CrossRef]
- Lu, X.-W.; Qin, S.; Wang, Q.; Lu, C.-Y.; Li, W.-J. Functional activity and mechanisms of common marine traditional Chinese medicine. Food Med. Homol. 2025, 2, 9420075. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Z.; Liu, W.; Ke, X.; Tian, X.; Chu, J. Cephalosporin C biosynthesis and fermentation in Acremonium chrysogenum. Appl. Microbiol. Biotechnol. 2022, 106, 6413–6426. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yao, X.; Yan, N. Structure of human Cav2.2 channel blocked by the painkiller ziconotide. Nature 2021, 596, 143–147. [Google Scholar] [CrossRef]
- Hossain, A.; Dave, D.; Shahidi, F. Northern sea cucumber (Cucumaria frondosa): A potential candidate for functional food, nutraceutical, and pharmaceutical sector. Mar. Drugs 2020, 18, 274. [Google Scholar] [CrossRef]
- Chen, L.; Du, S.; Qu, W.Y.; Guo, F.R.; Wang, G.Y. Biosynthetic potential of culturable bacteria associated with Apostichopus japonicus. J. Appl. Microbiol. 2019, 127, 1686–1697. [Google Scholar] [CrossRef]
- Leopold-Messer, S.; Chepkirui, C.; Mabesoone, M.F.J.; Meyer, J.; Paoli, L.; Sunagawa, S.; Uria, A.R.; Wakimoto, T.; Piel, J. Animal-associated marine Acidobacteria with a rich natural-product repertoire. Chem 2023, 9, 3696–3713. [Google Scholar] [CrossRef]
- Chen, L.; Wang, X.-Y.; Liu, R.-Z.; Wang, G.-Y. Culturable microorganisms associated with sea cucumbers and microbial natural products. Mar. Drugs 2021, 19, 461. [Google Scholar] [CrossRef]
- Tang, C.; Gao, R.; Tang, X.; Zhang, Y.; Feng, W.; Feng, B.; Lu, X. Metabolites isolated from Penicillium HDS-Z-1E, an endophytic fungal strain isolated from Taxus cuspidata and their activation effect of catalase. Chin. Herb. Med. 2024, 16, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Jiang, L.; Zhao, P.; Chen, H.; Jia, X.; Zhao, L.; Dai, H.; Hu, J.; Liu, C.; Shim, S.H.; et al. Chaetoglobosins and azaphilones from Chaetomium globosum associated with Apostichopus japonicus. Appl. Microbiol. Biotechnol. 2020, 104, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, W.; Lu, Y.; Wu, Y.; Ouyang, Z.; Tu, Y.; He, B. Epigenetic regulation of fungal secondary metabolism. J. Fungi 2024, 10, 648. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Tiwari, H.; Singh, S.; Gupta, P.; Rai, N.; Kumar Singh, S.; Singh, B.P.; Rao, S.; Gautam, V. Epigenetic manipulation for secondary metabolite activation in endophytic fungi: Current progress and future directions. Mycology 2023, 14, 275–291. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Pfannenstiel, B.T.; Keller, N.P. On top of biosynthetic gene clusters: How epigenetic machinery influences secondary metabolism in fungi. Biotechnol. Adv. 2019, 37, 107345. [Google Scholar] [CrossRef]
- Shwab, E.K.; Bok, J.W.; Tribus, M.; Galehr, J.; Graessle, S.; Keller, N.P. Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryot. Cell 2007, 6, 1656–1664. [Google Scholar] [CrossRef]
- Williams, R.B.; Henrikson, J.C.; Hoover, A.R.; Lee, A.E.; Cichewicz, R.H. Epigenetic remodeling of the fungal secondary metabolome. Org. Biomol. Chem. 2008, 6, 1895–1897. [Google Scholar] [CrossRef]
- Mao, X.-M.; Xu, W.; Li, D.; Yin, W.-B.; Chooi, Y.-H.; Li, Y.-Q.; Tang, Y.; Hu, Y. Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew. Chem. Int. Ed. Engl. 2015, 54, 7592–7596. [Google Scholar] [CrossRef]
- Guzman-Chavez, F.; Salo, O.; Samol, M.; Ries, M.; Kuipers, J.; Bovenberg, R.A.L.; Vreeken, R.J.; Driessen, A.J.M. Deregulation of secondary metabolism in a histone deacetylase mutant of Penicillium chrysogenum. Microbiologyopen 2018, 7, e00598. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Zhou, H.; Wang, X.; Huang, H.; Wang, H.; Zhang, R.; Wang, Z.; Han, J. Deletion of the histone deacetylase HdaA in endophytic fungus Penicillium chrysogenum Fes1701 induces the complex response of multiple bioactive secondary metabolite production and relevant gene cluster expression. Molecules 2020, 25, 3657. [Google Scholar] [CrossRef] [PubMed]
- Pidroni, A.; Faber, B.; Brosch, G.; Bauer, I.; Graessle, S. A class 1 histone deacetylase as major regulator of secondary metabolite production in Aspergillus nidulans. Front. Microbiol. 2018, 9, 2212. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Oberg, N.; Zallot, R.; Gerlt, J.A. EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) web resource for genomic enzymology tools. J. Mol. Biol. 2023, 435, 168018. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Nødvig, C.S.; Nielsen, J.B.; Kogle, M.E.; Mortensen, U.H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE 2015, 10, e0133085. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P.; Qi, C.; Zha, Z.; Meng, J.; Liu, C.; Han, J.; Zhou, Q.; Luo, Z.; Wang, J.; et al. Cryptic piperazine derivatives activated by knocking out the global regulator LaeA in Aspergillus flavipes. Bioorganic Med. Chem. 2024, 103, 117685. [Google Scholar] [CrossRef]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods. 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Deshpande, D.; Chhugani, K.; Chang, Y.; Karlsberg, A.; Loeffler, C.; Zhang, J.; Muszyńska, A.; Munteanu, V.; Yang, H.; Rotman, J.; et al. RNA-seq data science: From raw data to effective interpretation. Front. Genet. 2023, 14, 997383. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Zheng, J.Y.; Ren, S.N.; Yin, W.; Xia, X.; Li, Y.; Wang, H.L. A comprehensive workflow for optimizing RNA-seq data analysis. BMC Genom. 2024, 25, 631. [Google Scholar] [CrossRef] [PubMed]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 2022, 38, 2943–2945. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Maccurin, T.; Reilly, J. Sclerotiorine, a chlorinated metabolic product of Penicillium Sclerotiorum, Van Beyma. Nature 1940, 146, 335. [Google Scholar] [CrossRef]
- Arai, N.; Shiomi, K.; Tomoda, H.; Tabata, N.; Yang, D.J.; Masuma, R.; Kawakubo, T.; Ōmura, S. Isochromophilones III-VI, inhibitors of acyl-CoA:cholesterol acyltransferase produced by Penicillium multicolor FO-3216. J. Antibiot. 1995, 48, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Seto, H.; Tanabe, M. Utilization of 13C-13C coupling in structural and biosynthetic studies. III. Ochrephilone—A new fungal metabolite. Tetrahedron Lett. 1974, 15, 651–654. [Google Scholar] [CrossRef]
- Birkinshaw, J.H.; Kalyanpur, M.G.; Stickings, C.E. Studies in the biochemistry of micro-organisms. 113. Pencolide, a nitrogen-containing metabolite of Penicillium multicolor Grigorieva-Manilova and Poradielova. Biochem. J. 1963, 86, 237–243. [Google Scholar] [CrossRef]
- Fielding, H.C.; Graham, N.B.; Robertson, A.; Travers, R.B.; Whalley, W.B. 988. The chemistry of fungi. Part XXX. apoSclerotioramine and its derivatives. J. Chem. Soc. 1957, 4931–4944. [Google Scholar] [CrossRef]
- Bind, S.; Bind, S.; Sharma, A.K.; Chaturvedi, P. Epigenetic Modification: A Key Tool for Secondary Metabolite Production in Microorganisms. Front. Microbiol. 2022, 13, 784109. [Google Scholar] [CrossRef]
- Lee, I.; Oh, J.-H.; Keats Shwab, E.; Dagenais, T.R.T.; Andes, D.; Keller, N.P. HdaA, a class 2 histone deacetylase of Aspergillus fumigatus, affects germination and secondary metabolite production. Fungal Genet. Biol. 2009, 46, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhou, H.; Zhang, P.; Wang, X.; Li, W.; Zhang, W.; Liu, X.; Liu, H.-W.; Keller, N.P.; An, Z.; et al. Polyketide production of pestaloficiols and macrodiolide ficiolides revealed by manipulations of epigenetic regulators in an endophytic fungus. Org. Lett. 2016, 18, 1832–1835. [Google Scholar] [CrossRef]
- Akiyama, D.Y.; Rocha, M.C.; Costa, J.H.; Teles, C.B.; da Silva Zuccoli, G.; Malavazi, I.; Fill, T.P. The Penicillium brasilianum histone deacetylase clr3 regulates secondary metabolite production and tolerance to oxidative stress. J. Fungi 2022, 8, 514. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Cao, S.; Wang, J.; Lin, J.; Zhang, Y.; Liu, C.; Liu, H.; Zhang, Q.; Wang, M.; Meng, Y.; et al. Activation of secondary metabolite gene clusters in Chaetomium olivaceum via the deletion of a histone deacetylase. Appl. Microbiol. Biotechnol. 2024, 108, 332. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Wang, P.; Yu, Q.; Wang, B.; Li, D.; Yang, C.; Liu, L.; Duan, G.; Sun, W. The histone deacetylase UvHOS2 regulates vegetative growth, conidiation, ustilaginoidin synthesis, and pathogenicity in Ustilaginoidea virens. Phytopathol. Res. 2024, 6, 11. [Google Scholar] [CrossRef]
- Ma, H.; Li, L.; Gai, Y.; Zhang, X.; Chen, Y.; Zhuo, X.; Cao, Y.; Jiao, C.; Gmitter, F.; Li, H. Histone acetyltransferases and deacetylases are required for virulence, conidiation, DNA damage repair, and multiple stresses resistance of Alternaria alternata. Front. Microbiol. 2021, 12, 783633. [Google Scholar] [CrossRef]
- Lan, H.; Wu, L.; Sun, R.; Keller, N.P.; Yang, K.; Ye, L.; He, S.; Zhang, F.; Wang, S. The HosA histone deacetylase regulates aflatoxin biosynthesis through direct regulation of aflatoxin cluster genes. Mol. Plant-Microbe Interact. 2019, 32, 1210–1228. [Google Scholar] [CrossRef]
- Pavesi, C.; Flon, V.; Mann, S.; Leleu, S.; Prado, S.; Franck, X. Biosynthesis of azaphilones: A review. Nat. Prod. Rep. 2021, 38, 1058–1071. [Google Scholar] [CrossRef]
- Chen, C.; Tao, H.; Chen, W.; Yang, B.; Zhou, X.; Luo, X.; Liu, Y. Recent advances in the chemistry and biology of azaphilones. RSC Adv. 2020, 10, 10197–10220. [Google Scholar] [CrossRef]
- Hemtasin, C.; Kanokmedhakul, S.; Moosophon, P.; Soytong, K.; Kanokmedhakul, K. Bioactive azaphilones from the fungus Penicillium multicolor CM01. Phytochem. Lett. 2016, 16, 56–60. [Google Scholar] [CrossRef]
- Chiang, Y.-M.; Szewczyk, E.; Davidson, A.D.; Keller, N.; Oakley, B.R.; Wang, C.C.C. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J. Am. Chem. Soc. 2009, 131, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, W.; Tang, S.; Wei, S.; Lu, X. Collaborative biosynthesis of a class of bioactive azaphilones by two separate gene clusters containing four PKS/NPPSs with transcriptional crosstalk in fungi. Angew. Chem. Int. Ed. Engl. 2020, 59, 4349–4353. [Google Scholar] [CrossRef]
- Winter, J.M.; Sato, M.; Sugimoto, S.; Chiou, G.; Garg, N.K.; Tang, Y.; Watanabe, K. Identification and characterization of the chaetoviridin and chaetomugilin gene cluster in Chaetomium globosum reveal dual functions of an iterative highly-reducing polyketide synthase. J. Am. Chem. Soc. 2012, 134, 17900–17903. [Google Scholar] [CrossRef] [PubMed]
- Zabala, A.O.; Xu, W.; Chooi, Y.-H.; Tang, Y. Characterization of a silent azaphilone gene cluster from Aspergillus niger ATCC 1015 reveals a hydroxylation-mediated pyran-ring formation. Chem. Biol. 2012, 19, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-W.; Li, C.-Y.; Chiang, Y.-M.; Lin, T.-S.; Warren, S.; Chang, F.-R.; Wang, C.C.C. Characterization of a silent azaphilone biosynthesis gene cluster in Aspergillus terreus NIH 2624. Fungal Genet. Biol. 2022, 160, 103694. [Google Scholar] [CrossRef]
- McMullin, R.M.; Wing, S.R.; Sabadel, A.J.M.; Hageman, K.J. Experimentally derived biochemical modelling parameters to improve understanding of aquaculture’s effect on marine food webs. Mar. Environ. Res. 2022, 178, 105645. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, P.; Lin, J.; Zhang, Q.; Zhang, T.; Zhu, G.; Liu, C.; Wu, Q.; Qi, J.; Li, M.; Zhang, L.; et al. Deletion of the Class 1 Histone Deacetylase PsHos2 Induces Secondary Metabolic Perturbations in the Sea Cucumber-Associated Penicillium sclerotiorum. Fermentation 2025, 11, 230. https://doi.org/10.3390/fermentation11040230
Zhao P, Lin J, Zhang Q, Zhang T, Zhu G, Liu C, Wu Q, Qi J, Li M, Zhang L, et al. Deletion of the Class 1 Histone Deacetylase PsHos2 Induces Secondary Metabolic Perturbations in the Sea Cucumber-Associated Penicillium sclerotiorum. Fermentation. 2025; 11(4):230. https://doi.org/10.3390/fermentation11040230
Chicago/Turabian StyleZhao, Peipei, Jiaying Lin, Qingqing Zhang, Tanghui Zhang, Guoliang Zhu, Chengwei Liu, Qinghua Wu, Jianzhao Qi, Minglei Li, Lixin Zhang, and et al. 2025. "Deletion of the Class 1 Histone Deacetylase PsHos2 Induces Secondary Metabolic Perturbations in the Sea Cucumber-Associated Penicillium sclerotiorum" Fermentation 11, no. 4: 230. https://doi.org/10.3390/fermentation11040230
APA StyleZhao, P., Lin, J., Zhang, Q., Zhang, T., Zhu, G., Liu, C., Wu, Q., Qi, J., Li, M., Zhang, L., & Xia, X. (2025). Deletion of the Class 1 Histone Deacetylase PsHos2 Induces Secondary Metabolic Perturbations in the Sea Cucumber-Associated Penicillium sclerotiorum. Fermentation, 11(4), 230. https://doi.org/10.3390/fermentation11040230