Abstract
The dependence of photochemistry on excitation wavelength is not a recently observed phenomenon; nonetheless, it has, surprisingly enough, been largely ignored in the field. The reasons for this situation are not fully understood but might be related to a provisional extension of Kasha’s rule to photochemistry, or perhaps to a difficulty to justify the kind of short time-scales implied in such photochemistry, that challenges the usually held view giving predominance to fast internal conversion and vibrational relaxation. Regardless of the reasons, it is still a matter of fact that a complete and satisfactory interpretation for experimentally proven wavelength-dependent photochemistry is not yet available and the community endeavor to build a holistic understanding and a comprehensive view of the phenomenon. The present review is a non-exhaustive overview of the published data in the field, reporting on some of the most prominent features, issues, and interpretations.
1. Introduction
Despite the impact of excitation wavelength on photochemical reactivity ( effect), which has been acknowledged since the mid-twentieth century (around 1950), only recently have the effects of activation wavelengths become of genuine interest [1]. It has been argued that experimental observation relative to wavelength-dependent photochemistry has remained fragmentary and the regularities of the phenomenon are still neither well understood nor predictable [2,3]. Nonetheless, the literature advocates that the reactions originating from higher excited state are much more common than anticipated [4,5].
Much of the published work denotes such phenomena as anti-Kasha, in reference to Kasha’s rule [6], which is supposed to have pre-eminence in this area. However, as we shall see, this denomination (i.e., anti-Kasha) might not be appropriate because Kasha’s rule does not necessarily apply to photochemical events [7]. A complete interpretation of irradiation-wavelength variation in photoreactivity is not yet available but one can separate the observed data into roughly two groups. The first one corresponds to irradiation at different electronic absorption bands of the reactant, whereas the second concerns sequential irradiation, at narrow wavelength-intervals, of a single absorption band of the reactant. The former type of irradiation would readily involve different excited states, whereas the latter most likely operates within the same excited state. The reactions’ time-scales involved in each might differ. The former might invoke competition of photochemistry with internal conversion (), while reactions induced by the latter type of irradiation would proceed at an even shorter timescale (i.e., competing with vibrational relaxation ()). This analysis indicates rather a fast-to-ultrafast photochemistry, with time spans of 10−11 s (for ) or shorter [8], which falls beyond the time frame corresponding to luminescence (~10−9 s or longer), and hence does not corroborate an interpretation that suggests the applicability of Kasha’s rule to photochemistry.
Evidence of the variation in photoactivity with irradiation wavelength implies the measurement of the photochemical quantum yield as one of, if not the best, metric for this purpose. A change in quantum yield values with irradiation necessarily requires experimental/reaction data to evidence the occurrence of the phenomenon. Alternatives to experimentally determined quantum yield values are not available, since theoretical calculation of photochemical quantum yields is a rather nascent field of research. We shall see that different experimental ways of determining the quantum yield have been adopted in different studies. A fact that underlines a lacking consensus in the community on this particular matter.
This review reports on some of the most prominent results on the variation in photochemical processes (i.e., composition of end products, photoreactivity, and/or change in the quantum yield values) when subjected to different irradiation lights. The selected cases presented in this review, despite their limited number, strongly support the genuine occurrence of excitation wavelength-dependent photochemistry.
2. The Historical Kasha’s Rule
In 1950, Kasha published his seminal paper describing emissive and possible stages of deactivation of a light-excited molecule. Later, the work became known as Kasha’s rule [6]. The theoretical derivation, according to Kasha’s rule, can be summarized by the classical Jablonski diagram [9,10]. After a photon absorption, the excited molecule tends, in a first step, to lose some of its excitation energy by vibrational relaxation (), e.g., , that occurs between any higher vibrational state () of the nth singlet excited states ( where ) and the lowest vibrational state of that excited state. The new molecular state will further lose energy by internal conversion () and () to ultimately reach the first excited state (as ). From that first () excited singlet state, the molecule can subsequently deactivate to (i) the singlet ground state () via a non-radiative process, (ii) by emitting a photon of light (, fluorescence), or (iii) proceeds to a change of spin multiplicity by an “intersystem crossing” () leading it to a triplet state ( → ). In the latter state, the molecule relaxes by to , then to the first excited triplet state (), and from there can either phosphoresce () or relax, through a non-radiative to the ground state (), (Figure 1).
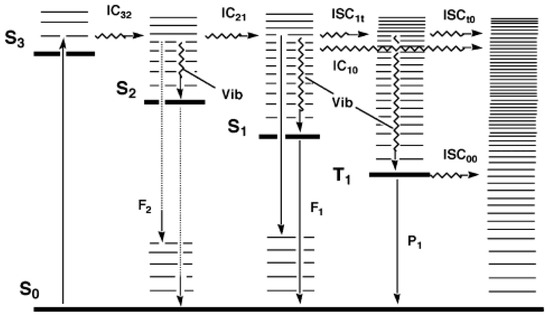
Figure 1.
Possible radiative and nonradiative processes of an organic molecule after absorption of a photon (). The non-radiative indicate internal conversions, whereas , , and , correspond to intersystem crossings from to , to , and to , respectively. , , and indicate radiative processes (fluorescence and phosphorescence) from , , and , respectively, and is the vibrational relaxation () in singlet and triplet manifolds. This modified Jablonski diagram includes an example of a non-Kasha emission () as an radiative transition. Reprinted with permission from [11]. 2024, ACS.
As observed from such photophysical processes, Kasha’s rule can be stated as “the emitting electronic level of a given multiplicity is the lowest excited level of that multiplicity ( or )” [6,12], but has also been stated as follows: “in solution, emission occurs from or , independently of the state which is initially excited” ( or ) [13,14]. This rule is also known as Kasha–Vavilov’s rule, with Vavilov’s rule stating that “the fluorescence quantum yield is independent of the excitation wavelength” [13,14,15].
The concept captured by Kasha–Vavilov’s rule is generally valid and practically observed in a large majority of luminescence experiments, but exceptions do exist as indicated by Figure 1 and stressed out, for instance, by the IUPAC’s “glossary of terms used in photochemistry” [14], as shall be further discussed below.
3. The Unfounded Extension of Kasha’s Rule Application to Photochemistry
Although, both Kasha’s and Vavilov’s rules were originally deemed to apply to molecular photophysics in condensed media, and neither author suggested their applicability beyond that context [7], it was recurrently considered that the application of Kasha’s rule “naturally” extends to photochemistry [3,12,16,17]. In substance, Kasha’s rule is thought to also stipulate that photoreactions only proceed from the same molecular energy levels from which emissions normally arise. According to this assumption, for any system, and are the only energetic levels from which molecular transformations, reactions, and measurable chemical changes will arise (since Kasha’s rule predicts that irrespective of the irradiation wavelength (i.e., excitation to ), the molecule will quickly decay to the lowest vibrational level of the first-excited electronic state (), from which it will be able to undergo photophysical processes, with this lowest excited state of the molecule presumably, also, representing the starting point for photochemistry).
There is enough experimental evidence in the photophysics literature to suggest that the emitted-light energy (e.g., the emission wavelength maximum), by either fluorescence or phosphorescence, is generally not disturbed by varying the excitation wavelength (as long as the remaining measurement conditions are kept unchanged). Such a simple experiment provides strong evidence supporting the argument that, for a majority of emitting species, fluorescence emission naturally originates from the lowest vibrational state of the first excited singlet state (). It is also the best indication of the validity of Kasha’s rule in photophysics for a large group of molecules. Exceptions to this norm will be reviewed in the following section.
Let us now consider the likelihood of an eventual extension of Kasha’s rule to photochemistry. Such an extension might be thought coherent with the view considering that within polychromatic molecules, rotational and even vibrational levels in reactants may be so “crowded” with the populations present at the ambient temperature that selection or excitation of individual levels is impossible [18].
In this context, it is perhaps also useful to observe that no such simple experiment, equivalent to that available to photophysics, (i.e., proving the invariance of the emission maximum with excitation wavelength), exists today to provide such irrefutable evidence on the occurrence of photochemistry from the lowest excited energy levels irrespective of excitation. For instance, such a straightforward experiment, if it existed, and led to a wavelength invariant photoreaction quantum yield, it would indicate Kasha’s rule applicability to photochemistry. It is important to stress here that supplying such an experimental proof, on the invariance in the photochemical reactivity with excitation, remains key to backing up an extension to and validating Kasha’s rule for photochemistry.
From a qualitative viewpoint, and to keep the spirit of Kasha’s rule for photochemistry, photochemical reactions should most likely proceed from (for singlet state photoreactions and from for triplet state reactions), and so, photochemistry must compete with photophysical luminescent processes (fluorescence or phosphorescence). If we assume that the above hypothesis stands, the time span of photochemistry has to be at least equal to that of fluorescence (~10−9 ) for singlet reactions proceeding from , and obviously much longer for the long-lived triplet state, . In this framework, there is difficulty in conciliating such timescales with those emerging from many time-resolved experiments that assign picosecond and even shorter femtosecond (10 to 100 fs) time spans for various molecular photoreactions [19,20,21].
Therefore, how to interpret this body of data? One can suggest that photochemistry seems to be well accommodated in sub-nanosecond timescales, not directly competing with fluorescence (as would be recommended by the hypothesis of an applicable Kasha’s rule to photochemistry). One is then forced to conclude that photochemistry is a much faster process than fluorescence for those cases, and that it belongs to a different time domain (much shorter than that generally assigned to fluorescence and phosphorescence).
Within this framework, the photochemical transformation of such molecules in their excited states takes places well before fluorescence. The observed reactant emission is then procured by photochemically unreactive species that relax to the first single-state (the proportion of excited molecules that did not photoreact). For proposed triplet state fast reactions, we can postulate a similar pathway, where photoreaction proceeds well before the emergence of phosphorescence that, if occurring, would be due to unreactive species. In this hypothesis, the intersystem crossing, leading to the formation of the triplet state, seem to effectively be a much faster process than supposed by the timescales measured for phosphorescence, as was documented for some molecules [22,23]. However, generally, the data available on triplet photoreactions tend to show relatively slow kinetics.
An interpretation of such phenomena can perhaps, at least in part, be provided by the conical intersection process. However, it is a matter of fact that there is no holistic explanation available today that is able to rationally connect the complete set of the available data.
Overall, the above qualitative analysis, which is undoubtedly a valuable hint, needs however to be supported by tangible experimental data. One of the ways to approach this problem is to look into the effect of varying excitation on the photochemical quantum yield (which is only determined experimentally). Indeed, according to an extended Kasha’s rule scheme, a given reaction is expected to proceed from a unique excited state (let it be the singlet state, ), and therefore the photochemical quantum yield of a given reaction step, such as , should not be affected by the excitation wavelength, i.e., whatever the excitation wavelength, the electrons first relax down to the first excited single state, and only then the photoreaction can proceed. Either way, experimentally testing the invariance of the quantum yield is a good approach to bringing evidence in support or against a supposed validity of Kasha’s rule for photochemistry. In essence, if the quantum yield is constant irrespective of the excitation wavelength, Kasha’s rule stands (as for photophysical processes); otherwise, photochemistry has to be considered faster than emission pathways, such as fluorescence.
4. Anti-Kasha Emission
Before we look into fast photochemistry, let us ask whether fluorescence may compete with or and whether such a behavior has been experimentally observed.
Within Kasha’s rule remit, photochemistry competes with fluorescence, and therefore, if fluorescence cannot compete with or , then photochemistry may well only proceed from the lowest vibrational level of the first excited singlet state (). However, if emission can proceed from either a higher electronic excited state (e.g., ) or a vibronic state (e.g., ), as suggested by Figure 1 (e.g., the F2 arrow), a possibility then opens for speculation on whether photochemistry is allowed for from higher energy excited states or from vibrational levels.
The so-called anti-Kasha emission is a terminology relating to a molecular emission of light that does not obey Kasha’s rule. That is, different excitation wavelengths induce emission spectra with distinct features. For a while, excitation wavelength-dependent fluorescence emission spectra were thought to be rather rare. However, the number of research groups claiming to have observed anti-Kasha emissions has become relatively large, even if the reported cases represent a relatively small fraction of light emitters, for which Kasha’s rule stands.
The phenomenon of wavelength-dependent fluorescence has been reported since the mid-1950s. Thiophosgene () is a particularly interesting case that exhibits, in the gas phase, an fluorescence that depends on the excitation wavelength [7,24,25]. Fluorescence emission was selectively observed from excitation of this molecule into the three first vibronic levels of , but not from higher vibronic levels where a fast internal conversion to the ground state, , occurs. The authors suggested that the rate of internal conversion was dependent on the vibronic levels reached upon excitation. The peculiar fluorescence of thiophosgene hence proves two extra possibilities for emission rates in addition to the usual : emission may compete not only with but also with . It is one of the first fluorescence emission cases that undeniably proved a dual limitation of Kasha’s rule for photophysical processes [26]. Thiophosgene emits from the second electronic excited singlet state (not from ), and this emission originates from different vibrational levels of , i.e., , , and . Similarly, in the gas phase, methyl salycilate [27], and three of its derivatives, methyl-2-hydroxy-3-naphthoate, methyl-1-hydroxy-2-naphthoate, and methyl-2-hydroxy-1-naphthoate, exhibited a variation in fluorescence quantum yield with the excitation wavelength [28].
Azulene [29,30,31,32], pyrene [33,34], benzopyran [35], and some of their derivatives are notoriously known examples of fluorescence in gas and condensed phases. Pyrene has been reported to emit from three singlet states, (,), (,), and (,) [33,36,37]. Historically, these molecules were thought to be exceptional anomalies, but more and more examples of wavelength-dependent fluorescence in condensed media were reported in subsequent years to enrich the documentation in this area [26,38,39].
Similar to the behavior of thiophosgene, rotationally cooled but electronically excited m-xylyl radical showed, in the gas phase, visible vibronic emission spectra originating from the transition [38]. Several thiocarbonyl compounds showed fluorescence, from singlet in solution [38]. In toluene, meso-tetraphenylporphyrin and other meso-substituted porphyrin derivatives have revealed a dual fluorescence emission process. The first emission was attributed to the usual transition, whereas the second emission process was assigned to electronic transition at the band, following excited-state tautomerism of the inner macrocyclic hydrogen atoms via an atom tunnelling process [39].
Interestingly enough, the anti-Kasha phenomenon of organic molecules in condensed media has been reported in two relatively recent reviews. One covering publications up to 2011 [11], and the other reporting on articles published between 2011 and 2021 [40]. They described excitation wavelength-dependent emission by experimental data and quantum chemical computational calculations for a wide variety of organic molecules, including azulenes, aromatic acenes, polyenes, thioketones, metalloporphyrins, aromatic carbonyl compounds, quinones, halogenated aromatic compounds, cyclic triimidazoles, 1,2-diphenylphenanthroimidazole derivatives, cyanines, thiophenes, and carbazoles.
The authors outlined the circumstances favorable for the occurrence of anti-Kasha’s rule behavior in condensed media. Mainly, two mechanisms were proposed to explain a fluorescence emission from : Either (ia) when both the oscillator strength () of the transition and the energy gap between and are large, or (ib) when of transition and the energy gap are very small, but with a concomitant large of the transition. However, fluorescence from higher () electronic levels was assigned to a relatively large energy gap between the electronic levels, combined to a prohibited internal conversion between and (due to symetry reasons). Phosphorescence anti-Kasha emission from (with ) was attributed to either (iia) an efficient to , whose symetry differs from that of , or (iib) to a thermal population of from .
It is perhaps also useful to mention here some red-edge effect examples. The so-called “red-edge effects” were related to the existence of excited state distribution of fluorophores in regard to their interaction energy with the environment and the slow rate of dielectric relaxation of this environment (hence, red-edge emission is not really anti-Kasha). The spectra were observed to be shifted to shorter wavelengths at the “blue” edge of the emission spectrum [41]. However, because of a usual relatively small spectral variation, this effect is not extensively investigated.
In a polar solvent, a giant “red-edge effect” was observed on the fluorescence of graphene oxide, which was strongly dependent on the excitation wavelength [42]. The increase in the excitation wavelength value caused an up to 140 nm bathochromic shift in the fluorescence maximum of several graphene oxide sheets (e.g., measured values were 350/440 and 500/580), and a broadening in the emission spectra. An and singlet transitions were assigned to the emission of an -rich graphene oxide derivative. Conversely, none of these features, including the excitation wavelength effect, was observed for graphene derivatives when a non-polar solvent was used [42].
The fluorescence spectra of many aromatic fluorophores embedded into different rigid and highly viscous media can depend on excitation wavelength, and the excited-state energy transfer, if present, fails at the “red” excitation edge [41,43,44,45,46]. Static inhomogeneous broadening was also observed for phosphorescence spectra, where it causes long-wavelength shifts in phosphorescence maxima at the red-edge excitation in motion-restricting media [47,48,49].
An excitation wavelength-dependent room-temperature phosphorescence was also reported in liquid and solid samples [50,51]. For instance, the color tunability of a series of phosphinoamines (phenylbenzothiazoles bearing phosphine groups [50]), in both solid state and solution, was due to a dual-band emission of hundreds of microseconds lifespan whose intensity depends on the excitation wavelength of ultraviolet light. DFT calculations supported an interpretation for such a nonconventional radiative process by involving a combination of pathways, including a locally excited state (the transition between excited states with different molecular geometry), a charge transfer state, and/or an excited-state intramolecular proton transfer.
5. Distribution of the Literature in Photochemistry
It seems appropriate, for the purposes of the present review, to divide the literature relative to the possible dependence of photochemistry on excitation into two groups: One encompasses those publications where the authors considered that Kasha’s rule applies to photoreactions without providing further discussion about the matter, or experimentally investigating its validity. One can stipulate that this first group of research considers Kasha’s rule as a principle for photoreactions. Because the latter is not yet proven with certainty, we will not be including such studies here.
The second type of documentation relates to research papers that sought to obtain experimental facts on whether the excitation wavelength has an impact on photoreactivity and/or on the values of the photochemical quantum yield. Some of these will be reviewed here. Of particular interest is the literature that employed monochromatic beams for quantum yield measurements, since polychromatic light has an averaging effect that might not allow for critically resolving differences between individual wavelength quantum yield values. Accordingly, it seems important to particularly review the literature that provides tangible experimental evidence on the effects of varying the monochromatic irradiation wavelength on the absolute values of the photochemical quantum yield of photoreactions. In this context, one must acknowledge a variability in the methods and equations used for the determination of the quantum yield even when monochromatic light is employed.
6. Early Observations
Within less than three decades from Kasha’s publication [6], Turro and his co-workers [7] compiled what perhaps represents the first comprehensive review on the effects of excitation wavelength on photoreactions. It covered a substantial amount of experimental data (more than 180 papers over the 1955–1977 period), spanning a series of phototransformations in molecules belonging to a variety of chemical families and involving an array of different photomechanisms.
Most of the described compounds showed a variation in the composition of the photoproducts when the irradiation wavelength, impinging on the reactive medium, was changed, i.e., different sets of end-products were generated under irradiation with different excitation wavelengths (whose beams were mostly provided by unfiltered or filtered lights of various lamps). Contrastingly, only a few studies reported a difference of the quantum yield with irradiation wavelength but with no alteration in the photoproduct set generated by the reaction.
The alteration in the reaction outcome in response to a change in the excitation wavelength (-effects) was attributed to phenomena arising from either excited state or ground state properties of the reactive species. The authors of the review proposed to rank this type of reaction into four classes: (a) photoreactions for which the wavelength dependence is due to the ground state properties only; (b) photoreactions starting from higher electronic excited states (), or ; (c) photoreactions starting from higher vibrational states ((hot) excited states indicated by the symbol ) of the lowest excited electronic states, i.e., or ; and (d) reactions from through the population of the latter state from or . The review focused more on differentiating spurious -effects due to ground state properties from -effects implying higher excited state or vibronic photochemistry than with assigning the observed reactions to the latter two categories. The authors have, however, attempted to account for reactions with fast or slow internal conversion features.
The four classes listed above turned out to, a priori, be sufficient to explain the cases reported in the review [7], and make a solid ground for a general classification.
7. Photoreactions of Species in Equilibrium
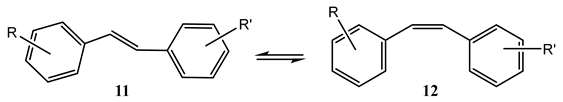
Here, a rapid thermal equilibrium is envisaged between the starting materials. A pertinent situation whenever a relatively modest thermal barrier (not exceeding ~17 kcal/mol [7]) exists between the interconvertible molecular structures (e.g., ). In this instance, we are dealing with ground state species ( and , where is a product of ), obtained from thermal equilibration (Scheme 1). In general, the irradiation of the solution may concern two or, concomitantly, several species in thermal equilibrium. In these circumstances, different excited states are populated by the excitation (e.g., and ).
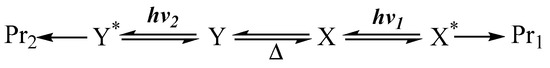
Scheme 1.
Photoreactions of species in thermal equilibrium.
Each excited species might undergo an independent primary photoreaction, so that, when subjected to different irradiation wavelengths ( and ), the photoreaction of one species might yield a different set of end-products (e.g., ) to those (e.g., ) obtained from another species (Scheme 1).
Another mechanism that is overall analogous to that presented in Scheme 1 corresponds to the situation where a photostationary state, pss, (instead of the thermal equilibration) is established between and . Here, the reactant () is first exposed to a light, , until pss is reached. Then, lights of different wavelengths ( and , where might be equal to ) are shone on the solution (now containing both thermally stable and ). Each irradiation light will favour one end-product (Scheme 2). This mechanism can also concern the central photoreversible reaction only (in which case and ).
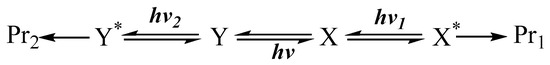
Scheme 2.
Independent end-product sets due to differences in the excitation wavelength of the lights driving a reactive system in its photostationsry state.
In the general case, the electronic absorption spectra of species and might or might not overlap, but they are likely to have different absorptivities. The accumulation of and in the solution will hence depend on both their respective production quantum yields ( and ), absorptivities ( and , assuming that and do not, themselves, absorb) at the irradiation wavelengths, and the irradiation time or the intensities of the incident light beams. In these conditions, such processes, as given in Scheme 1 and Scheme 2, can readily be explained without invoking the features of the excited states. Indeed, if one of the species is characterized by either or at the irradiation wavelength, and/or when the ratio is very small or very high, then only the other species will be observed in solution (or at least in the largest proportion).
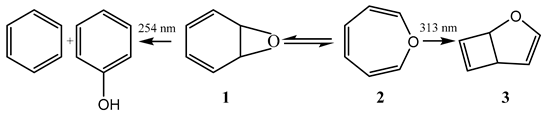
The benzene oxide reaction is an example of the mechanism provided by Scheme 1. Exposure of an ether solution of benzene oxide (1) at −80 °C to a 254 nm light resulted in the formation of benzene and phenol. However, at room temperature under a beam of light of wavelength higher than 310 nm, only (3) was obtained [7,52]. The authors explained these results by the occurrence of a thermal equilibrium between (1) and (2). At low temperature, the equilibrium strongly favors (1), and hence only the reaction of that species is observed, whereas at room temperature, where (1) and (2) coexist, the reaction of (2) is predominant because its absorptivity for wavelengths > 310 nm greatly exceeds that of (1).
Bicyclo[4,3,0] nona-2,4-diene] (4) irradiation, in pentane, with a 300 nm light beam leads to (6), whereas its exposure to a 254 nm light leads to (5) [53]. In fact, a pss is established between (4) and (5), with this ring closure process ((4)→(6)) being much faster than that of the ring opening ((4)→(5)). This means that over a very long exposure time with either irradiation wavelengths, only (6) is expected to remain in solution.
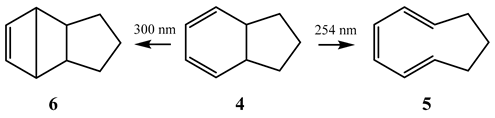
The experimental observation of a wavelength dependence of the end-product of (4) for relatively short exposure times has been linked to the relative absorptivities of the three species at each irradiation wavelength. At 254 nm, the ratio , but at 300 nm, it drops to ~0.02. Therefore, the reaction undertaken at 300 nm favors the formation of the less efficient (6), whereas at 254 nm, the pss is displaced towards (5). A valid interpretation of the experimental observation without invoking excited states.
Even though we observe a measurable effect of the irradiation wavelength, Kasha’s rule still cannot be tested by the systems shown in Scheme 1 and Scheme 2, due to the occurrence of a hypothetical quasi-independent reaction of from that of . The photoreactions of and might or might not, separately, obey Kasha’s rule. The experiment using such systems (Scheme 1 and Scheme 2) cannot provide the useful information to reach an answer if only one irradiation is performed. Therefore, to identify whether wavelength has an impact on the reactivity of each individual species ( and ), it is required to perform a systematic screening of the sample under several irradiation wavelengths and the data relative to each individual species treated separately.
The earliest known example of such a screening is that of azobenzene photoisomerization [54]. The wavelength dependence of the photoreversible azobenzene forward and reverse quantum yields was measured at 254, 313, 365, 405, 436, 546, and 578 nm. The trans (7) to cis (8) quantum yield almost doubled, ranging from 0.13 to 0.27, respectively, between 254 and 436 nm but decreases between 436 and 578 nm to a final value of 0.23. The cis-trans (8 → 7) efficiencies showed a zig-zag pattern in the region 254–578 nm varying in the 0.40–0.55 range.
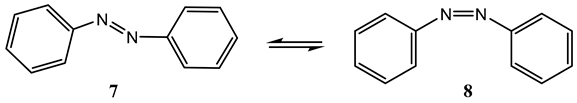
Several subsequent studies have confirmed both the variability in azobenzene quantum yield with excitation wavelength and the lower trans-cis quantum yield value compared to that of its counterpart reverse reaction (Table 1) [55,56]. Lower quantum yields of azobenzene were also reported for shorter UV wavelengths [55,57]. The latter results were corroborated by a multiconfiguration calculation of azobenzene along the NN bond twisting, which identified an singlet state of the trans geometry (as the lowest excited singlet state), on the basis of the doubly excited configuration n2π*2 [58]. The authors found that the latter state provides an explanation for azobenzene quantum yield wavelength dependence.

Table 1.
Photoisomerization quantum yields of azobenzene in various solvents and under different irradiation wavelengths (mercury lamp lines). Reprinted with permission from [56], 2024, Taylor & Francis.
The quantum yields of azobenzene isomers were determined experimentally on the basis of a differential equation for the rate processes that assumes monochromatic light, vigorous stirring, the validity of Beer–Lambert’s law, and that the quantum yields in both directions are independent of both the light flux provided to the reaction medium and the initial concentration [54]. The non-linear rate law was simplified by discarding a term involving the thermal reaction rate constant, and its linearization was achieved by expanding the photokinetic factor in power series for high concentrations (~10−4 to 10−3 M). The trans-cis quantum yield was, hence, obtained from the gradient of the linear relationship of the light flux with time. This information facilitated the subsequent determination of the cis → trans quantum yield from the steady-state equation. It is also interesting to remark that a substantial decrease in the photoisomerization rate with increasing initial concentration was shown in the study. The variation in the quantum yield with wavelength was confirmed later in methanol by numerically solving the system of the differential equation of the reaction and the mass balance for the forward and reverse quantum yield values [59].
Solving for the quantum yields of photoreversible systems such as azobenzene, diarylethenes, spiropyrans, and fulgides through using numerical integration was also recommended [60,61]. This contrasts with the approaches calculating the quantum yield of photoreversible systems such as azobenzenes and stilbenes, whereby the formulae of each quantum yield were worked out from a mathematical method using the equations corresponding to irradiations at two different wavelengths but assuming that the quantum yield is wavelength-independent [62]. The latter strategy of evaluating the quantum yield using numerical calculation and irradiation at multiple wavelengths while considering an invariant quantum yield was also adopted for photochromes’ photokinetic data such as spiropyrans and spirooxazines [63,64]. Unfortunately, such a mathematical strategy is not very helpful since the validity of Kasha’s rule for photochemistry is, de facto, accepted and translated in the equations.
An attempt to approximate the wavelength-dependent quantum yield values of both isomers was performed by the formula , where is the excess vibrational energy immediately after internal conversion, is the height of the potential energy barrier, and the number of classically excited modes of vibration [54]. It was found that this model not only predicts a monotonic decrease in quantum yield with increasing excitation wavelength, but the quantum yield estimated values were less than 0.01 for all wavelengths. Both characteristics are not conforming with their experimental measurements. They also concluded that the latter findings are not consistent with previously proposed models by Lewis [65] and Olson [66] for cis-trans isomerization. Furthermore, in the case of azobenzene, it was observed that the experimental data do not point to an indistinguishable common intermediate state, regardless of which isomer, trans or cis, was excited by the radiation, because the sum of the individual quantum yields, measured at each wavelength, do not add up to 1 [67,68], i.e., at a given irradiation wavelength. For the authors, the results on the quantum yield dependence of wavelength strongly suggested the existence of two separate configurations of the excited states ( and ) of the two individual isomers trans and cis, according to Scheme 3.
Scheme 3.
A photoreaction mechanism proposing independent and azobenzene excited states (but excluding the occurrence of a common intermediate).
The latter hypothesis was somewhat corroborated by a CASSCF computation of the excited-state relaxed surfaces of trans-azobenzene, which indicates that the photoisomerization mechanism can both proceed by rotation from a cold species and by a concerted inversion when excitation in produces a hot species. These two excited-state species may explain the much lower reaction quantum yield from than that observed from excitation [69]. Incidentally, a sigmoid-like trend in quantum yield vs. temperature was observed for trans-azobenzene-modified DNA [70].
DNA–psoralen monoadduct photoreaction quantum yield values ranged between 0.012 and 0.08 when the excitation varied between 248 and 365 nm, in a sort of a convex pattern (with 0.08 and 0.04 quantum yield values recorded at the short and long wavelengths of the curve) [71].
The light-driven cis (10) to trans (9) isomerization of the cis-retinal chromophore in the G-protein-coupled receptor rhodopsin was, for a long time, assumed to be wavelength-independent [72] but later, in a more accurate measurement [73,74], was found to be constant between 450 and 480 nm, and falls significantly as the irradiation wavelength increases to 500 nm ( 0.67). A further 5% reduction in the quantum yield values was obtained at 570 nm. The quantum yield value at 500 nm and its wavelength dependence was corroborated by calculation and experiment [75,76].
Kim and co-authors [73,74] hence corrected the long-held presumption that the quantum yield in vision was wavelength-independent and supported the hypothesis that the 200 fs photoisomerization reaction that initiates vision is dictated by nonstationary excited-state vibrational wave packet dynamics, which corroborates the previous conclusions of an earlier study on all-trans-retinal whose excitation-dependent fluorescence was assigned to a competition between photochemistry and internal conversion and between the vibrational levels of its first-excited singlet state [77]. The quantum yield of cis-retinal chromophore in the G-protein was calculated by Equation (1):
where is Avogadro’s number, is the incident radiation intensity, is the molar decadic extinction coefficient (M−1 cm−1) at the irradiation wavelength, is the optical path length (cm), is a correction factor due to other absorbing species, and is the Dartnall’s function of the absorbance () [78].
The findings of Kim and co-authors [73] were compared to the predictions provided by a Landau–Zener model for dynamic curve crossing [74]. The treatment of the nonstatistical internal conversion process revealed that absorption by unreactive high-frequency modes increases as the excitation wavelength shifts from 570 to 500 nm, whereas relatively less energy is deposited into reactive low-frequency modes (indicating the importance of delocalized, tortional modes in the reactive pathway of rhodopsin). However, the variation in the quantum yield of rhodopsin was assigned to a ground state heterogeneity of a mixture of cis and all-trans retinal absorbing around 480 and 540 nm [79] (but unfortunately no data from analytical chemistry techniques were shown to identify the ground state mixture).
Irradiation in different absorption bands of trans-4-nitro-4′-dimethylamino-stlibene (11) in cyclohexane was reported to produce (a) a little variation in the isomerization quantum yields (e.g., 0.16 and = 0.42), (b) a value well below unity for the sum of the individual forward and reverse reactions’ quantum yields, but, at the same time, (c) a significant change in the percentage of the cis-isomer in the pss composition, ranging between 19 and 64%, for irradiations performed, respectively, between 313 and 436 nm [80]. The study also demonstrated the occurrence of both temperature and concentration effects on the quantum yields. For instance, the of 4-nitro-3′-methoxy-stilbene in 1-methyl-naphthalene reduced from 0.32 to 0, as the concentration gradually increases from 10−5 M to 1 M.
A variation in the quantum yield of trans to cis stilbene in n-hexane was recorded between 313 nm (0.27) and 254 nm (0.47), whereas a relatively constant quantum yield (0.29) in the cis to trans reaction was measured for the same irradiation wavelengths [81]. The authors acknowledged differences in the way the quantum yields were determined in the individual different studies from which the wavelength effect was reported, and in general, a similar trend was observed for a number of stilbene derivatives.
In our team, systematic irradiations (typically at 10-to-20 nm intervals) were conducted on a variety of molecules undergoing E/Z photoisomerization. In ethanol solutions, the irradiation spanned the whole electronic absorption spectrum of the molecule investigated. The photoisomerization governing the reactions obeyed Scheme 2. The quantum yields were determined for the individual E- and Z-isomers, whose absorption spectra most often overlapped over a large section of wavelengths. The calculation of the individual quantum yield values was worked out by a semi-empirical photokinetic method based on the -order kinetics model [82,83]. The experimental temporal variation in the total absorbance of the medium , which is subjected to a monochromatic, non-isosbestic irradiation (at ) and monitored at the observation wavelength (), is fitted to the model equation, Equation (3).
where is the recorded total absorbance at long reaction time; the absorbances are measured at either the same () or different () wavelength of observation and wavelength of irradiation. and are the optical path lengths of the irradiation and monitoring lights inside the sample (in ), is the photoisomerization reaction rate constant (in ), is the time (), is the decimal logarithm, and the exponential function.
The quantum yields of the reactant (e.g., the trans-isomer) and the product (e.g., the cis-isomer) are then obtained from Equations (4) and (5).
where is the absorbance coefficient (in ) of the isomer species indicated, measured at either or ; is the initial reaction rate (in ) that is worked out by differentiation of Equation (3) at ; is the incident light flux per irradiated area and volume of the sample (in ); is the photokinetic factor at the initial time and at the end of the reaction; and is the initial reactant concentration (in ).
Except for the trans → cis photoisomerization of sunitinib (13) [84], the quantum yields for both E- and Z-isomers, including those of asthma drug montelukast (17) [85], the serotonin neuronal inhibitor fluvoxamine (15) [83], the orally active multiple tyrosine kinase and angiogenesis inhibitor, the cis-isomer of anti-cancer sunitinib [84], the antioxidant stilbenoid oxy-resveratrol (14) [86], and the Phytoalexin plants’ infection and stress protector pinosylvin (16) [87], all showed a clear variation in irradiation wavelength (Table 2).
Table 2.
Quantum yields patterns and span over the investigated irradiation wavelength sections of several molecules subjected to excitation with monochromatic light.
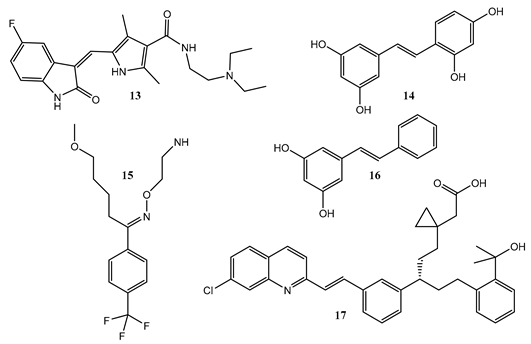
Several patterns characterized the variation of the quantum yield with wavelength, including sigmoid, linear, triangular, and curved shapes (Table 2). The sum of forward and reverse quantum yields never added up to unity (). Also, for all the studied molecules, the ratio () was higher or lower than unity depending on the molecule and its geometry. The data hence indicated that the cis and trans isomers behaved as independent species in the photoisomerization process.
The photokinetic model developed here has also provided an explicit formula for the reaction rate constant (, Equation (6)), which represents the first example of its type in the literature, for a photoreversible reaction whose isomers both absorb at the wavelength of the non-isosbestic monochromatic irradiation inducing the reaction [82].
It is obvious from Equation (6) that the reaction rate constant does not depend exclusively on the absorption coefficient of the reactant as might have been supposed, i.e., the reaction would be faster where the absorption is higher (the rate depends on absorption). In fact, the rate constant of the photoisomerization is a much more inclusive function involving all the parameters of the reaction at the irradiation wavelength (including the irradiated volume and area of the reactive medium, both present in the formula of [88,89]), meaning that the reaction rate constant is a relative quantity that can neither be related to a specific parameter (such as an absorption coefficient) nor evaluate the quantum yield of either forward or reverse reactions. However, Equation (6) indicates the fact that the ratio of the isomers’ concentrations at pss () might well vary with changing one or more of the reaction parameters including the irradiation wavelength, a feature that may explain many experimental observations of variable E/Z concentration ratios.
8. Photoreactions of Multi-Chromophoric Species Yielding Different End-Products
Some of the interpretations proposed in the previous section do not discard the fact that these reactions derive primarily from excited state processes. If features of the latter were not really required for the interpretation of some of those reactions, the excited state information might, however, be relevant to other situations.
For instance, and conversely to the previous cases involving two (or more) chemically independent species (Scheme 1 and Scheme 2), photochemistry can develop from a single reactant whose reactivity might well be dependent on the irradiation wavelength. The analysis now needs to rely on the excited state properties. In the simplest case (Scheme 4), two different end-products ( and ) are obtained by exposing the reactant to lights of two different wavelengths. The most likely interpretation adopted for this type of reaction considers the occurrence of two distinct excited states of (e.g., and ). The latter, non-equivalent excited states, may have either singlet or triplet character (, , …, , ,…etc.), generally of ,, ,, or , type. The model shown in Scheme 4 implicitly implies that photochemistry from the excited states and , must occur before redistribution of the excitation energy within the molecule [7,22].
Scheme 4.
A reactant photoconvesion yielding different sets of photoproducts under different irradiations conditions.
The most probative example of such a mechanism can stem from the excitation of different chromophores (e.g., and associated with the excited states or , respectively), both belonging to the molecular structure of . The specific reactions leading to the end-products and (or more) represent a tangible proof of the difference in the primary photoprocesses, presumably proceeding from and , respectively.
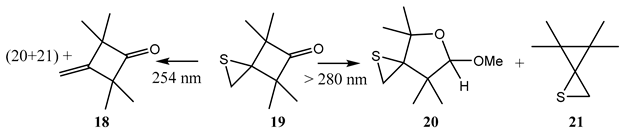
A typical—and one of the earliest—photoreactions of this type concerned molecules endowed with a spatially separated bond and a carbonyl group [90,91]. For the 4,4,6,6-tetramethyl-1-thiaspiro[2,3]hexan-5-one (19), in methanol and under a 280 nm light, the carbonyl is excited to an , singlet (), which results in the expected ring expansion to the acetals (20) and (21), without perturbation of the other cyclic moiety of the sulfur atom [91]. But a 254 nm beam (through quartz) specifically induced an , transition of the bond, corresponding to the molecular state, that leads to cyclobutanone (18) and the removal of the sulfur atom.
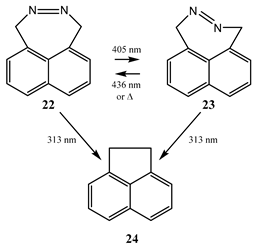
In ethanol at room temperature, a 405 nm irradiation of l,4-dihydronaphtho[1,8-de] [1,2] diazepine (22) [92,93] excites the N=N bond, corresponding to an , transition that exclusively gives the highly strained trans-isomer (23). The latter reconverts into (22) in the dark at room temperature, or by light excitation of its , band situated at ca. 436 nm. However, irradiation of the naphthalene absorption bands at ~ 313 nm, mainly populating the , states of the individual isomers (22) and (23), led to the exclusive formation of acenaphthene (24). The authors estimated the ratio of the cis → trans and trans → cis quantum yields to be 1/10, as derived from the wavelength dependence of the isomer pss compositions [22]/[23]. Notice that both (22) and (23) can individually be considered as the reactant () in Scheme 4. This kind of reaction is a process that was sometimes labelled chromatic orthogonality [94,95,96].
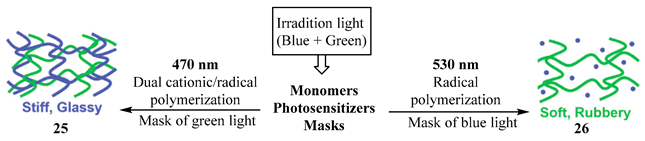
When polychromatic light is used on reactions of Scheme 4, such as when the two chromophores might be excited (e.g., (as an ,) and (as a ,)), selectivity can then be achieved by employing an absorption competitor whose absorption spectrum covers one of the electronic transitions (e.g., the , responsible of ), so that only the product generated from the other transition (e.g., the , of ) is obtained as the major (or the only) species in the solution. A rationalization of this phenomenon through -order kinetics has been proposed [89]. This approach has widely been used for molecules encompassing a variety of dual chromophores that were envisaged for various applications. Notably, adopted in chromatic orthogonality and exploited in releasing photolabile protecting groups [97,98,99,100,101,102,103,104,105,106,107,108]. As an illustration, a 3D printing process requires an irradiation consisting of both blue and green lights, which should alternatively be reduced to either green or blue by the use of an adequate absorber (mask) of the unwanted light color. The use of blue light irradiation induced both radical and cationic photopolymerization that produced the stiff, glassy material (25). Conversely, green light-absorbing photosensitizer led only to radical photopolymerization of acrylates, producing a soft, rubbery material (26). Based on the chemical nature of the different domains, the 3D printed multi-material object displayed interesting and tunable mechanical properties, including increased toughness and resistance to crack propagation compared to single component materials [107,109].
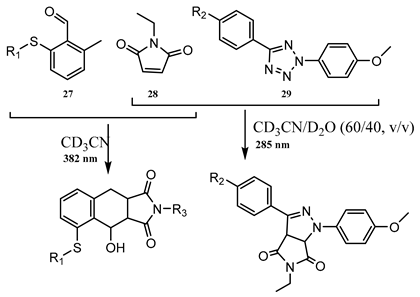
An analogous process of sequence-independent wavelength-selective (-orthogonal) photoreactivity can be achieved by a selective interplay between the absorption properties of the reactive species, the quantum yields of the separate reactions in different solvents, and the irradiation wavelength [106,110]. For instance, when o-methylbenzaldehyde thioether (27), N-ethylmaleimide (28), and methoxy-substituted diphenyltetrazole (29) are all present in the reaction medium, it was demonstrated that in specific solvents, different reactions are induced by LED lights of different wavelengths.
In terms of interpretation, in cases where and happen to be both singlet states, the process can eventually be explained by considering that vibrational relaxation within a given state can be fast, but not the internal conversion from higher to lower energy states, . In this perspective, the latter internal conversion must be slow enough for photochemistry to proceed from the individual states. Turro and co-workers [7] proposed an interpretation based on the theoretical expression of the rate of internal conversion () [111] from the vibrational level of an upper excited state () to a lower excited state (), that is given by the following:
with and being the electronic wave functions of and , respectively, and the nuclear kinetic energy operator. and are the nuclear wavefunctions for the appropriate vibrational levels of and , respectively. The electronic factor , results from a coupling of the electronic wave functions, and is the Franck–Condon factor between the two states.
Equation (7) clearly predicts that internal conversion may be slow if either or both and have small values. The former situation (poor ) applies when the excited states ( and ) are orthogonal, and the latter (low ) corresponds to a large energy gap between and . Even though this analysis is useful, a general, more extended description would most likely include non-adiabatic and spin-orbit coupling terms that may affect transfer among states.
9. Photoreactions of Multi-Chromophoric Species Yielding a Unique Product Irrespective of the Excitation Wavelength
One more situation (Scheme 5) embodies a photoconversion, with non-zero quantum yield, of one reactant to one set of end-products irrespective of the absorption band targeted by the irradiation.
Scheme 5.
A reactant phototransformation into a unique set of end-products irrespective of the irradiation wavelength used.
Photoreaction of a reactant may well be characterized by the selective reactivity from one excited state but not from the other [112]. The selectivity of a unique electronic excited state to channel reaction towards the end-product is easily identified in practice since it only requires performing irradiations at excitation wavelengths falling on the absorption band promoting the excited state channeling reaction.
In general, however, the reactions represented by Scheme 5 might mean that either there is a unique reaction channel irrespective of the excitation, such as when one of the excited states relaxes into the other (e.g., by internal conversion, or by then ), or the presumably different excited states relaxing down, through specific channels, to the same set of end-products.
It is reasonable to predict that for the former case, such a process should not, a priori, show any variation of the quantum yield when the excitation light takes different wavelengths that may induce excited states of different characters, e.g., ,, ,, or ,. If this hypothesis stands, it may be postulated that the reactive system is deemed compatible with Kasha’s rule, in the sense that the variation in irradiation wavelength does not affect the output (quantum yield) of the reaction due to the existence of a unique intermediate after excitation. This view will still hold when the irradiation is performed under polychromatic irradiation, since such a light usually covers more than one absorption band of the reactive molecule, an irradiation condition that is very often encountered in experimental photochemistry, as it predominantly uses lamps. In fact, this combination of reactions obeying Scheme 5 and irradiation using a lamp represents a non-negligible section of photoactive reactants. Therefore, the qualitative conclusion (since no quantum yields are measured) that might be advanced here is that this behavior fits well with Kasha’s rule (the end-product(s) is independent of the excitation wavelength). The ubiquity of such reactions and behavior in photochemistry may perhaps explain the propensity to adopt an extension of Kasha’s rule to photochemistry.
The second mechanistic case (multiple reaction channels) will rather accept the possibility of a difference of the quantum yields for different excitations owing to the supposed difference of the pathways leading to the end-product(s). However, and in any case, such hypotheses will only be validated if confirmed by a detailed experimental investigation of the irradiation wavelength effect on the quantum yield.
For the cases of the photoreversible systems presented in Table 2 (whose reactants individually belong to Scheme 5 reactions), they would rather demonstrate that Kasha’s rule does not apply, since the quantum yields of the individual photoisomers are variable with irradiation. In addition, for most of these molecules, the quantum yields recorded for reactions from states were higher than those staring from their higher excited states.
A short-wavelength excitation (at 254 nm, ) was found to enhance the photodegradation of estrone (30) under direct photolysis by individual beams in the 254–320 nm wavelength range [113].
Flash photolysis of a series of quinone derivatives (31) [114] was performed at four wavelengths (440, 400, 350, 260, and 220 nm, with the beams delivered through specific filters placed between the lamp and the sample). The selected wavelength set covers the major part of the absorption spectra of these compounds, which correspond to one large envelop spanning the 220–480 nm region. The study showed that except for the weak long-wavelength absorption band, a decrease in quantum yield values with decreasing wavelength was manifest. Irradiation of the shortest absorption band in ethanol (220 and 260 nm) was characterized by the lowest quantum yield values (0.01 to 0.1) compared to those (0.07 to 0.98) recorded for longer-wavelength (e.g., 350 nm) irradiations.
Several α,β-unsaturated γ-dimethoxymethyl cyclohexenones (32) were found to undergo certain unimolecular reactions when excited to the (,) state. The latter competes with internal conversion and intersystem crossing. The unimolecular reactions do not, however, occur from the (,) or the lowest triplet (, and ,) states. A very low quantum yield (<0.001) of (32) resulted from irradiation to its (,) state, compared to the quantum yield (0.02) recorded for its phototransformation from a , state into (33) [115].
This type of photoreaction is widely exploited in the photoremoval of protecting groups and photocages. The molecules combining a protecting group (such as coumarin (34)) covalently linked to bioactive compounds (i.e., the caged species, morphine (35)), are totally inactive biologically. Irradiation with poly- or monochromatic light removes the protecting group and uncages the bioactive molecule [116].
The photocleavage, leading to the production of an oxygen molecule, in a series of endoperoxides were found to exhibit a wavelength-dependent quantum yield. For instance, the quantum yields of (36) and (37) were constant (respectively, 0.13 and 0.18) between 290 and 313 nm ( excited state), but for the state populated with irradiation at 365 nm, the quantum yields reduced to less than 2% of the constant values recorded above (respectively, 0.0026 and 0.0015). Similarly, release from –superoxide compounds [117] depended on the excitation wavelength, recording a more than eightfold decrease between 436 nm (0.29) and 683 nm (0.035).
Dimethyldihydropyrene (trans-10b,10c-dimethyl-10b,10c-dihydropyrene, closed-form isomer) exhibits a negative photochromism, where the photoconversion of the colored (38) into the colorless, cyclophanediene, open-form isomer (39) is triggered by visible light. Nonetheless, the quantum yield of the forward (38 → 39) reaction has a low but positive value (0.08%) at 470 nm (corresponding to an state), whereas its value is zero for an excitation of the transition at 660 nm [118].
Overall, the reactions described, thus far, are transition-specific whether the result is a unique set of end-products for different excitation wavelengths, or the end-products are obtained for excitation of one band, but not for the excitation of other transitions. The description might also be considered relative to the quantum yield values, whereby, for the type of species (32), only for irradiation of a given absorption band, but for irradiation of the other bands.
The photodegradations of anti-cancer dacarbazine (40) and cardiovascular nifedipine (41) and nisoldipine drugs show a reverse behavior whereby irradiation of the short-wavelength bands ( state) translates into much less photoconversion than in irradiation of the long-wavelength absorption bands (-state) [119,120,121,122]. The systematic screening of the excitation wavelength effect on the reaction quantum yield showed a monotonical increase of , whose pattern fits a full sigmoid function for nifedipine and nisoldipine, and a partial one for dacarbazine phototransformations (Figure 2). Quantum yields were measured at short intervals over wavelength regions where both reactant and product absorbed the irradiation light, by using Equations (3)–(5) (with ), at low initial reactant concentration (~5 × 10−6 M), and under strictly monochromatic irradiation. The quantum yield values of the state reactions are relatively small (~10−3), being ca. 50 times smaller than those recorded for the drugs’ states.
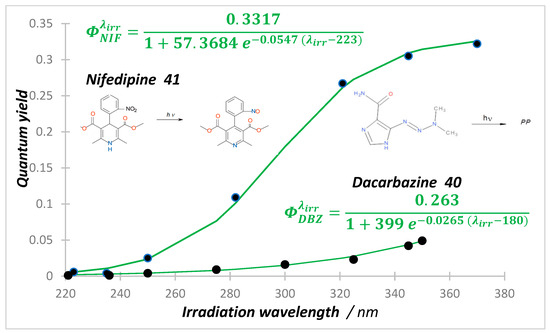
Figure 2.
Increase in quantum yield values of both nifedipine () and dacarbazine () with the increase in the monochromatic irradiation light wavelength (circles). Reactions performed in ethanol at 22 °C, with initial concentrations of M for DBZ (40) and M for NIF (41). The equations correspond to the sigmoid patterns (green solid lines). Adapted with permission from [120,122]. 2024, Elsevier.
The photostationary state (pss) composition and the quantum yields of photoisomers of a series of diarylethene-containing benzothiazolium species (42) were found to strongly vary with irradiation wavelength when using mercury lamps (365/405 nm and 405/436 nm) to induce the photoisomerization [123]. For this series, the shorter the wavelength, the lower the pss percentage of the Z-isomer, and were smaller than at different excitation wavelengths, which has discouraged postulating a common excited state intermediate.
The cyclization quantum yield of 1,2-bis(5-(4-ethynylphenyl)-2-methylthiophen-3-yl) perfluorocyclopentene was characterized by significantly higher values (>0.7) compared to those (<0.16) recorded for the ring-opening reaction [124]. The quantum yields were determined by the mathematical expression Equation (8).
with being the increase or decrease in absorbance at 600 nm recorded under exposure of the solution of volume to a light flux over the period , being the absorbance of the solution at the specific irradiation wavelength, and being the absorption coefficient at 600 nm. Both quantum yields showed dependence on the monochromatic irradiation wavelength. The ring-closing quantum yield varied between 0.71 and 0.92 in the UV range (334–254 nm) but was constant () between 334 and 313 nm, whereas that of ring opening showed a ten-times increase (from 0.0015 to 0.016) when visible-range irradiation decreased from 609 to 404 nm. A negative linear relationship was obtained in acetonitrile for the ring-opening quantum yield (). Even though the authors stated that no explanation can be advanced for the differences in quantum yields, they nonetheless estimated that such a behavior might be due to the existence of an excited state energy barrier for the closed-form isomer but not for its counterpart.
The ring closure reaction of a benzoyl–phenyl–ethinyl-substituted 1,2-bis [2-methyl-thien-3-yl] perfluorocyclopentene takes place with a high quantum yield through a conical intersection, which is also a relaxation funnel for the closed isomer -excited-state preceded by an energy barrier [125]. The closed form was proven to be a mixture of three isomers (cis-cis, cis-trans, and trans-trans) with different quantum yields. For solutions of the diarylethene in (5 × 10−4 to 5 × 10−5 M), the quantum yields were wavelength-dependent, varying between 0.9 × 10−5 and 2.5 × 10−4 for irradiation wavelengths situated in the 514–690 nm region. The quantum yield of the ring-closing reaction was reported as being dependent on both excitation wavelength and the doses of irradiation.
The choice of the excitation wavelength (297, 313, and 334 nm) has no effect on the quantum yield (~0.03) of ring-opening of perfluoro- and perhydroxycyclopentane diarylethene derivatives in acetonitrile (~2 × 10−5 M at 25 °C), whereas under the same wavelength series, a 15% variation for the ring-closure (0.51–0.60) reaction quantum yield was recorded [126]. The quantum yields were determined at non-isosbestic wavelengths located in a region where the isomers’ electronic spectra overlap. They were calculated by a method exempt of simplifying assumption using pure photokinetic data, requiring the irradiation to be collimated and monochromatic but free from imposing any other special irradiation conditions [127].
The quantum yield of the ring-opening reaction (, corresponding to 43 → 42) of photochromic diarylethene derivative (l,2-bis [2-methylbenzo[b]-thiophen-3,3,4,4,5,5-hexafluoro-1-cyclopentene) was measured spectrophotokinetically by Equation (9).
This formula is the only known example in photochemistry that is analytically derived, by a closed-form integration, from the rate law describing a reaction subjected to non-isosbestic irradiation [128]. The absence of approximations and/or simplifying assumptions in the elaboration of Equation (9), as well as its obvious difference to the analytically derived equations for the thermal kinetics (i.e., zero-, first-, and second-order reactions), has promoted this equation to becoming the representative of the new -order kinetics. Irradiation of an organic solution of the colored isomer (43) in the visible region of the electromagnetic spectrum (where both the solvent and the open-form isomer (42) do not absorb), reveals a clearly dependent quantum yield on the excitation wavelength. In ethanol and under monochromatic irradiation over the 410–535 nm range, the quantum yield of the ring-opening reaction follows for a distinctive sigmoid pattern, Figure 3 [129]. A linear relationship () was also obtained for this derivative in acetonitrile [130].
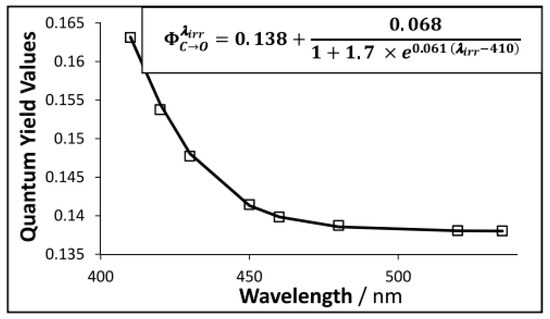
Figure 3.
Experimentally measured quantum yield () values of the closed- to open-form isomers reaction of a diarylethene derivative (squares) subjected to strictly monochromatic irradiation beams covering the whole absorption spectrum of the closed-form isomer in the visible region (3.39 × 10−6 M in ethanol at 22 °C). The sigmoid pattern (solid line) is given by the inset equation. Reprinted with permission from [129]. 2024, Elsevier.
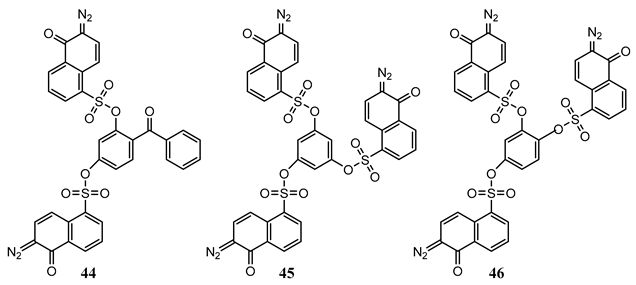
Three derivatives, (44) to (46), bearing two or three chromophoric diazonaphthoquinone units were investigated in acetonitrile (ca. 2 × 10−5 M) under various monochromatic radiations at 254, 350, 399, and 423 nm at 290 and 80 K. It was found that the photoconversion quantum yields, measured by two different methods, varied between 11 and 55% for the individual molecules over the selected series of irradiation wavelengths [131].
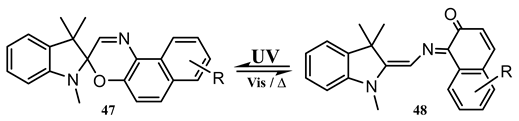
By irradiating the transition of a spirooxazine derivative (47), 1,3-dihydro-3,3-dimethyl-1-isobutyl-6′-(2,3-dihydro-1H-indol-1-yl)spiro(2H-indole-2,3′-3H-naphthop [2,1-b] [1,4] oxazine), it was observed that the quantum yield of the photoconversion decreased with wavelength (0.22 and 0.24 at 410 and 355 nm, respectively), whereas upon excitation to a higher state (), the quantum yield value first decreased to 0.1 at 316 nm then increased to 0.53 at 295 nm (a V-shape pattern). These findings were interpreted as the photochemical yield being mainly determined by the vibrational mode of the excited singlet state where the molecule is prepared [132].
The ratios of photobleaching and photocoloration quantum yields of a series of spironaphthoxazines [133] were unequivocally proven to depend on the excitation wavelength at 352 and 414 nm on the basis of using a modified Fischer’s method [134]. (That is, in this study, the expressions calculating the individual quantum yields were obtained from a Fisher’s method assuming that the ratio of bleaching/coloration quantum yields was constant, and that the rates of the thermal reactions of ring opening were not negligible).
The pss compositions as well as the quantum yields of a series of water-soluble spiropyrans (49) were shown to be excitation wavelength-sensitive. The ring-opening (49 → 50) reaction efficiencies underwent between 26 and 96% decreases for, respectively, irradiations occurring at 260 and 365 nm. A mixed increase and decrease in the quantum yield values at 530 nm was observed for different molecules [135].
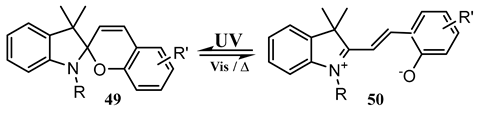
The (at 260 nm) excitation results in a more efficient reaction pathway than the (at 350 nm) excitation. The quantum yield was calculated by the ratio of the number of molecules that actually underwent phototransformation () to the number of photons absorbed by those molecules (), as
The photocoloration quantum yield of an oligothiophene-substituted naphthopyran derivative decreased from 0.32 to 0.045 when monochromatic and continuous irradiation increased from 318 to 410 nm [136]. Similarly, the photochemical study of a biphotochromic naphthopyran was carried out at diluted (6 × 10−6 M) and highly (3 × 10−3 M) concentrated toluene solutions [137]. The quantum yield decreased by a third (from 0.32 to 0.11) with increasing wavelength of monochromatic beams spanning the 330–410 nm range, where measurements were performed at a 10 nm interval. Integration, initial rate, and differential methods were used for the estimation of the quantum yield at each irradiation wavelength. The dependence of the reaction quantum yield on wavelength within the excitation band, for both studies, was thought to indicate that the photoreaction occurs at a vibrational excited level in competition with vibrational relaxation.
Photoswitchable fulgides also represent a family of interest from the point of view of quantum yield variation with excitation. The experimental results showed, in general, that the quantum yield of the open- to closed-form isomer was slightly lower than that corresponding to the ring-opening quantum yield, and both recorded higher values to the reaction induced by visible light ( over 500 nm). These values varied between 0.15 and 0.20 (at ca. 400 nm, for ring closing), ranged between 0.1 and 0.26 (in the region 300 and 400 nm, for ring opening), and between 0.03 and 0.07 for irradiation in the visible range (500 to 600 nm) [138,139,140,141]. The data of quantum yield values measured at 15–20 nm intervals over the 340–700 nm spectral region served the proposal of a trifluoromethyl–indolyl–fulgide derivative as a UV-to-NIR actinometer [142]. The quantum yield was calculated by Equation (11) for concentrated solutions exposed to monochromatic beams (where the absorption of the reactive medium at the irradiation wavelength was ~ 3).
where is the molar amount (in mol) of the photo-species that reacted in time , is the molar amount (in einstein) of photons absorbed, and is the photon flux (in einstein s−1) of the light source that is absorbed by the sample.
The quantum yield of the ring-closing reaction was attributed to the (52 → 53) reaction step as a spectral unmixing of the irradiation series showed only a minor content of the E-isomer in the reactive medium. The proposed mechanism included independent photoisomerization of the excited state species into their isomers ( and ) as well as a thermal equilibrium of the ground state isomers (). The latter process has been neglected for this particular molecule since both its forms were found thermally stable, i.e., this reaction mechanism involves only photochemical reaction steps.
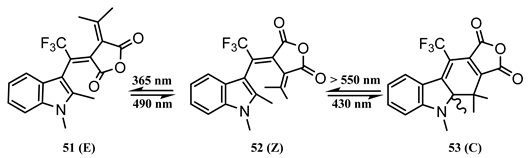
Substantial photochemical production of the -isomer as well as thermal reactions between the ground state species have been observed for a number of fulgide derivatives [143,144]. These features render the determination of the specific quantum yield values for each photoreaction step of the reaction mechanism a tedious task, a consideration that must raise caution in regard to the quantum yield values reported in the literature for fulgides.
In general, quantum yields are determined for thermally stable reactants but not for every photochemical step of the reaction mechanism leading to a photostationary state (e.g., fulgides (51) to (53)). In this context, it is useful to mention an unprecedented set of kinetic formulae that were developed for the concentrations of a reactant and its four photoproducts, sequentially generated in a multi-consecutive photoreaction [145]. These -order kinetics formulae allowed for determining the quantum yields of riboflavin (54) phototransformation into (55), which itself subsequently photoconverts into (56).
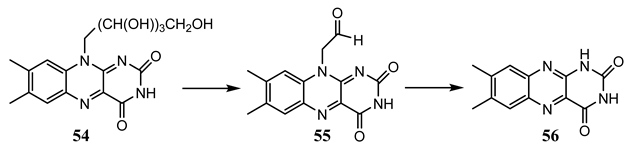
Ethanolic solutions of (54) of initial concentrations ca. 10−6 M, were exposed to monochromatic collimated light beams of wavelengths 400, 420, 445, 460, and 480 nm, and the individual quantum yield of (54) and (55) at each irradiation wavelength were determined (Figure 4). For both species, the quantum yield values increased with irradiation wavelength. A more pronounced variation was observed for (55) with a 5-fold increment between the lowest and highest recorded quantum yields compared to only a 1.5-fold variation for the quantum yields corresponding to (54). The quantum yield values for (54) were 6- to 20-times higher than those of (55), but both were well described by sigmoid functions. The wavelength range used here (400–480 nm) corresponds to two absorption bands (380–410 nm and 420–490 nm, respectively); hence, excitation occurs both in different states ( and ) and at different wavelengths of the lower energy state, .
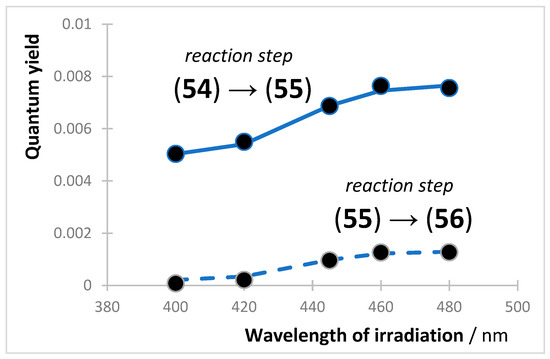
Figure 4.
Sigmoid variation in the experimental photochemical quantum yield of the reactions (54) → (55) (top) and (55) → (56) (bottom), in ethanol at 22 °C. Reprinted with permission from [146]. 2024, Elsevier.
10. Vibronic States Photochemistry
Becker and his co-worker were perhaps the first to lay down, in the late 1960s [146], an interpretation directly linking photochemistry to vibronic states. The fluorescence excitation and absorption spectra of 2,2-diethylchromene were found to be very different [147], and the quantum yields of fluorescence () of this compound as well as those of 5,6-benzochromene showed up to a 10-fold variation with the particular nature of which the state was excited, including the electronic state, the vibrational mode, and the vibrational level. The photoconversion of such molecules seemed to compete with fluorescence emission but not with phosphorescence (which was not observed, and the estimated quantum yield of was low ~0.1 [148,149]). A kinetic model (Equation (12)) was proposed for the fluorescence quantum yield as a result of a competition between vibration relaxation (with a rate constant ) and photoreaction (with a rate constant ) at the excited vibrational level, [146].
The analysis was later carried forward [150] to establish an expression (Equation (13)) for the photoreaction quantum yield in relation to features of both photophysical and photochemical processes, in addition to, and, for the first time in the field, taking into account a quantum yield of vibrational relaxation of a given excited vibrational level within a given mode of any electronic excited state (Figure 5).
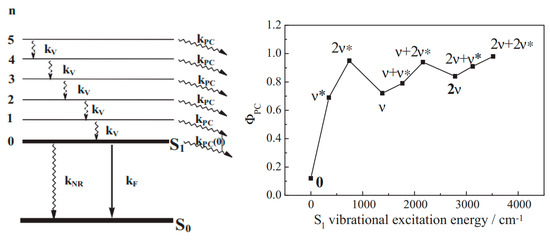
Figure 5.
(Left): Representation of possible photophysical and photochemical processes in the first excited state with the rate constants for from to (), (), fluorescence (), and photoconversion (). (Right): Illustration of the variation in the photoconversion quantum yield of a non-fluorescent photochrome when excited at different vibrational levels of the first electronic excited state. Reprinted with permission from [151]. 2024, Elsevier.
It was concluded that photochemistry ought to be competitive with vibrational relaxation at any excited level (electronic or vibrational/vibronic). A number of subsequent studies of various molecular structures has led to similar correlations between photochemical quantum yield values and excited vibrational levels, as was described in a detailed review [151]. It has been noted that when polychromatic light is used, the determined quantum yield of a given reaction is rather an average yield over several wavelengths and, therefore, the published data on quantum yield should be considered with caution. It is also worth mentioning that the model proposed by Becker and his co-workers always predicts higher quantum yield values for shorter excitation wavelengths due to the additive nature of the model. The latter point is shared with the model, discussed above in Section 7 [54], that predicts a monotonic decrease in the quantum yield with increasing excitation wavelength. As such, these models do not seem suitable for many observed cases where the quantum yield value increased with wavelength.
A recent systematic review and analysis of reactions involving intersystem crossing, photoisomerization, bond-breaking, and electron, proton, and energy transfer, proved the competition of photoreaction with , radiative, and non-radiative processes occurring in upper excited states [5]. Analysis of experimental data suggested that both and to proceed by slower or comparable rates to those of photoconversion. Direct coupling of specific vibrations and photoconversion has been observed in many experiments [152,153], and vibrational coherence has been detected for isomerization [154], electron transfer [155,156], and proton transfer [157]. A reversed V-shaped variation of the quantum yield with excitation wavelength (525, 550, 585, 625, and 600 nm) was recorded for 6-methyl-azulene-2-carboxilic acid where the quantum yield value steeply decreased from ~0.8 at 525 nm to ~0.2 at 585 nm then gradually to ~0.1 at 600 nm [158].
The data presented in Table 2 and Figure 2, Figure 3 and Figure 4 also indicate the occurrence of vibronic photochemistry since variation in quantum yield was recorded when the state was subjected to irradiation at a series of different wavelengths.
Evidence of the contribution of vibrational levels to photochemistry has been laid out for many other systems. The small chlorine dioxide () molecule undergoes, in solution, vibrational mode-dependent photochemistry [159,160,161].
An interesting review focused on some photochemical reactions in the atmosphere that are driven not only by UV light but also by low-energy visible light, as observed at high solar zenith angles [162]. Absorbed near-IR and visible region photons, promoting stretching vibrational overtones, contribute to atmospheric chemistry and play a role in the fate of certain organic compounds [163,164,165,166]. Three mechanisms were envisaged for these photoreactions. The excited overtone redistributes its vibrational energy within the molecules, so that enough energy is transferred into a different weaker bond of the molecule that ultimately breaks [167,168]. A second pathway involves a concerted reaction whereby a strong coupling of the excited overtone with the reaction coordinate leads to the excitation of a low-frequency mode, causing the bond to break at much lower energies than the dissociation energy of the bond at its equilibrium geometry [164,169]. Thirdly, a two-photon absorption sequentially brings the overtone to an intermediate state from which the system is promoted to a repulsive dissociative first excited electronic state [170]. Similarly, the initial vibrational state preparation was found to strongly influence the photodissociation of by excitation of an stretching overtone that enhances the breaking of the bond in its electronically excited state [171].
A vibrational mode-dependent excited state lifetime was investigated for phenol under the conical intersection, where the bond rupture dynamics were interpreted by a possible topographical link to a lower energy minimum on the multidimensional potential energy surface [172].
Formaldehyde () photolysis, dependent on the excitation wavelength and the variation of the quantum yield of the radical channel (Figure 6), was modelled by an expression (Equation (14)) involving a sum of sigmoid-like functions [173,174]. The excitation of vibrational states for this molecule is implicitly suggested from the narrow wavelength intervals used for irradiation (~5 nm) and the continuous variation in measured quantum yield values.
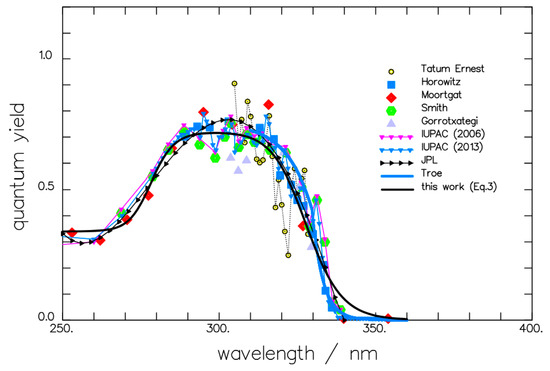
Figure 6.
Experimental variation in the radical channel quantum yield of formaldehyde photolysis at room temperature, modelled by Equation (14). Reprinted with permission from [174].
11. Some Inorganic Actinometers
The variation in quantum yield with excitation has also been reported for inorganic systems including actinometers. Two illustration examples are shown here, where the reported data correspond to photonic yields rather than absolute quantum yield values. The patterns observed are different but surely convey an effect of excitation.
Nitrate and nitrite photoreactions are monitored by the photochemical productions of salicylic acid (Figure 7a) and p-hydroxybenzoic acid, by the reaction of the photoproduced hydroxyl radical with benzoic acid [175].
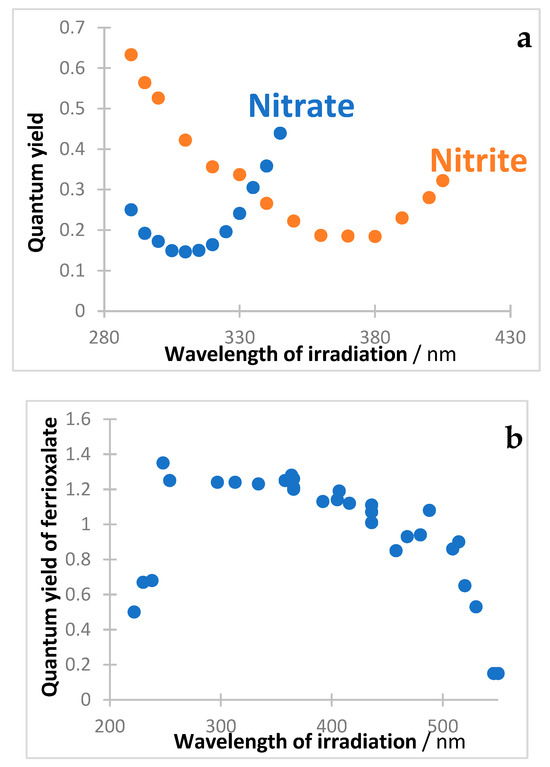
Figure 7.
(a) Interesting patterns of salicylic acid production quantum yields with irradiation wavelengths, from the photoreaction of either nitrate or nitrites. Raw data taken from [175]. (b) Evolution of the photochemical quantum yield of Fe3+ → Fe2+ with irradiation wavelength in the ferrioxalate actinometer. Raw data taken from [56].
Also, a series of studies revisited the properties of the ferrioxalate photoreaction that was originally proposed for chemical actinometry in 1956 [176]. The latter inspiring work established the wavelength sensitivity of the quantum yield of the ferric/ferrous ions photoconversion. A compilation of quantum yield values from several studies, gathered in the handbook of photochemistry [56], clearly shows that the photoreduction quantum yield of the ferrioxalate ferric ions evolves within its dynamic 200–550 nm range, in a similar pattern to that observed for formaldehyde photolysis (Figure 6), but with a majority of the values exceeding 1 (Figure 7b). The high quantum yield values (exceeding unity) were attributed to the occurrence of thermal processes.
12. Challenges/Limitations
There are cases where reactivity variation was recorded but quantum yields were not generally measured. The reactivity changes were due to changes in reaction/reactant features. Examples are found in industrial and technological applications. In this context, recent discussions on the quantum yield definitions and their respective equations differentiated between the experimentally measured quantities and their actual interpretation [88,89]. It is useful to remember that the quantum yield, a quantity measured at a given wavelength using strictly monochromatic light, is insensitive to reaction conditions such as initial concentration, radiation intensity, temperature, and absorption competitors. However, the photonic yield (also labelled photonic efficiency or apparent quantum yield) is a relative quantity that is necessarily sensitive to a variation in such reaction conditions, and the only one obtained when the employed irradiation light is polychromatic. The relative character of the photonic yield meant that it is possible to observe its variation with some reaction conditions (e.g., medium temperature), while it may remain unaffected by a change of the excitation wavelength, as was observed for photoreactions of both 9- and 9,10-bromoantracenes [177].
Despite light-based stimulus having gained noticeable popularity in drug delivery systems, long exposure to light remains a negative feature that might lead to phototoxicity (such mutations, degradation of endogenous molecules, photocarcinogenesis, etc.). In this field, it has been recognized that the wavelength of the light employed is one of the important factors to reduce toxicity to acceptable levels or to suppress it altogether [178]. The sensitivity of DNA as a function of the irradiation wavelength (corresponding to the sun’s spectrum reaching the surface of the earth) smoothly diminished, as a sigmoid pattern, between 260 and 360 nm [179]. The data were used to express the carcinogenic effectiveness of the light as a function of the light’s wavelength, and also to deduce a quantitative evaluation of the dose response in relation to UV-induced skin cancer.
In photocatalysis, semiconductor photocatalysts are increasingly characterized by the variation in photonic yield with excitation wavelength (the so-called action spectrum), which ideally has the same shape as that of the fraction of radiation absorbed by the semiconductor photocatalyst with wavelength [180,181,182]. The action spectrum indicates a clear correlation between irradiation wavelength and photoreactivity. Similarly, photocurrent action spectra were used in order to study the influence of the geometrical structure with respect to the efficiency of thin film devices [183].
Also, reaction/reactant features effects have long been observed on both forward and reverse quantum yields of photoreversible systems that depended on several reaction parameters such as viscosity, temperature, additives, initial concentration, and excitation-wavelength [81].
For instance, direct photolysis of polychlorinated biphenyls (PCB), in neutral 2-propanol at a series of irradiation wavelengths (254, 264, 294, and 300 nm) in a Rayonet photoreactor, showed a wavelength dependence on the apparent de-chlorination quantum yield (0.15 to 0.56) [184]. The apparent quantum yield was also found to increase with increasing the initial concentration of the PCB or the intensity of the irradiation light. The findings of the study were proposed as a template for the remediation of PCBs. Similarly, photoproduct formation (apparent) quantum yields of a series of fluorinated pesticides and pharmaceuticals were evaluated using LEDs emitting at 255, 275, 308, 365, and 405 nm. The fastest degradations were observed for irradiations with 255 or 275 nm compared to the other LEDs, and a different set of end-products was obtained with 365 nm irradiation [185].
Another interesting aspect in this area relates to the quantification of the quantum yield variation with structural changes in the molecule studied (e.g., in a series of derivatives), and with the consequent changes occurring in the ground state absorption features of the individual derivatives.
For instance, the photoinduced quantum yields of ring closure and ring-opening reactions of a series of 1,2-dithienylperfluorocyclopentene derivatives followed a general trend where a lowering of the derivative transition energy led to a decrease in the quantum yield of the photoreaction of both open and closed isomers, as well an increase in the quantum yield with increasing temperature [186]. The ring-opening reaction quantum yield increased roughly exponentially with increasing transition energy. A similar behavior was also observed for a series of dithienylethenes with polyenic and carotenoid-like chromophores, where the quantum yields of the photochromic forward and back reactions sharply decreased with increasing polyenic chain length [187]. Such a behavior was rationalized by the decrease in excitation density at the central photoreactive unit and the concomitant shortening in the excited state lifetime in the carotenoid compounds with increasing chain length. Also, an analogous dramatic decrease in ring-opening quantum yields in a series of thiophene oligomers (including up to three thiophene rings), was observed when increasing the number of thiophene rings, whereas only a little variation was recorded for the ring closure reaction [188]. The efficiency of the photoreactions was correlated to a threshold of the energy.
A quantification of such an effect was delivered for a series of six bis(thienyl)ethenes, studied in THF organic solutions and in PMMA films under irradiation from lasers or LED lights [189]. The quantum yield of the photocoloring reaction was determined specifically at the isosbestic point of each derivative (located in the region 300–330 nm), and those of the photobleaching reactions were measured by irradiation at the maximum of each colored isomer (in the region 574–605 nm). These quantum yields were determined by a spectrophotokinetic method [127,190] based on an analytical integration of the rate law of the reaction subjected to a monochromatic beam whose wavelength exactly corresponds to that of an isosbestic point. A plot of the isosbestic point wavelength against the corresponding quantum yield value for the bis(thienyl)ethene series shows good correlation as a sigmoid function (Figure 8). It is an interesting structure/photoactivity feature.
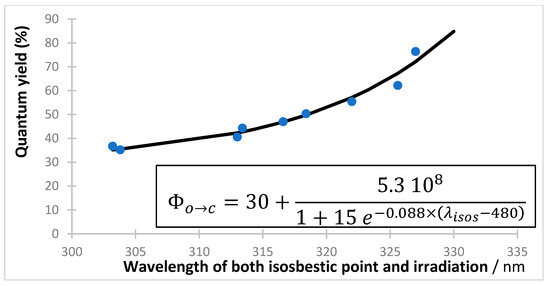
Figure 8.
Good sigmoid correlation (line) of the isosbestic point wavelength and the quantum yield values (dots, in %) at that wavelength for a series of bis(thienyl)ethene derivatives. The sigmoid function is indicated in the inset. Raw data taken from and adapted with permission from [189].
Photoproduced diarylethene isomers are generally thermally stable, but a few exceptions were reported. The ring-opening reaction of hexafluoro- and hexahydro-diarylethene derivatives, in organic solutions, was strongly temperature-dependent [191]. The thermal rates at different temperatures were worked out from the initial linear portion of a logarithmic scale plot of the kinetic data. The observed rate of the photoinduced ring-opening was dependent on radiation intensity but not the exponential component of its fitted traces. The authors attributed this feature to the presence of an excited state thermal barrier.
13. Concluding Remarks
The number of different chemical families exhibiting a photochemical quantum yield variation with excitation wavelength is arguably in support of an underlining fundamental process, and rather challenges the view considering such variation as an exotic or rare phenomenon. A focus on the examination of the occurrence of such a phenomenon is useful and should be recommended as an integral part of future studies.
The cases displayed in this review make it obvious that the conjecture stating that the reaction quantum yield must be constant irrespective of the excitation wavelength is proven false by the abundant literature. Evidence of reactions proceeding from higher excited states and/or from hot excited states has been mounting since the 1950s. The same has also been established for photophysical radiative processes. Competition of both luminescence and photochemistry with and/or opens new avenues of research and offers the possibility of selecting conditions suitable to specific needs and applications. As a result, Kasha’s rule does not necessarily always hold for all emitters and is not automatically extendable to photochemistry. Accordingly, there is need to amend the Jablonski diagram in line with the recurrent findings in the fields of photophysics and photochemistry to include such diversity in the possible pathways open to the excited molecule. Based on the data available, Figure 9 attempts to capture this complexity albeit in a synoptic view.
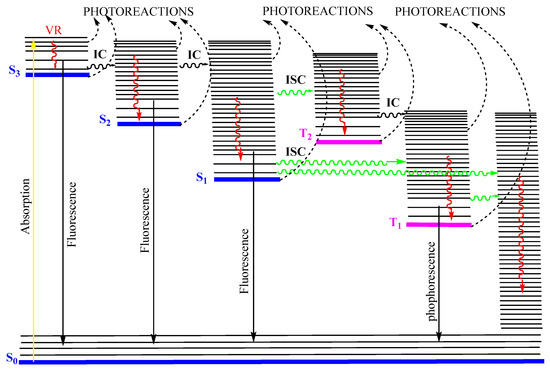
Figure 9.
An amended Jablonski diagram with pathways for photophysics and photochemistry emerging from hot and cold excited states conveying possible competition of these processes with internal conversion and/or vibrational relaxation.
The community agrees, on one hand, that experimentation is the only way to reveal which pattern will be taken by photoreactivity when changing the excitation wavelength (the prediction of such a pattern is not yet reliably accessible by any other means). The best metric for that purpose undoubtedly remains a measure of the quantum yield. Here, a distinction must be made between the quantum yield and the photonic yield. The former is an intrinsic feature of the molecule at a given wavelength that can only be determined by using a strictly monochromatic light, whereas the latter is a relative quantity that can also be obtained for photoreactions subjected to polychromatic light. The photonic yield varies with the change in experimental conditions of the reaction.
On the other hand, however, there is an obvious lack of consensus in the community on the standard ways, formulae, and photokinetic methods by which the quantum yield of a photoreaction is experimentally determined. This is shown by the large number of approaches that have been adopted for the evaluation of the reaction quantum yield (a sample of expressions and methods has been provided in this review). Standardization, hence, becomes necessary; it is an issue that needs to be addressed by the community in order to strengthen reliability of the data, and to improve the quality of the conclusions that can be reached for the effects of excitation on reactivity.
In this context, the data available in the literature on the effect of excitation wavelength most often relate to a few (two or three) measurements, whereas a systematic investigation over wider wavelength ranges and a larger number of actual measurements (preferably covering the whole absorption spectrum of the species studied, with measurements taken at narrow wavelength intervals) will provide a better understanding of the reaction quantum yield behavior (as monotonical increase or decrease, or as a variable change with increasing irradiation wavelength).
Finally, a full explanation of the quantum yield dependence on wavelength is not yet available despite the many attempts to describe it. This is a future task that will certainly reveal new subtilities of the excited state.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in the original papers cited in the text.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Kislyak, A.; Frisch, H.; Gernhardt, M.; Van Steenberge, P.H.M.; D’Hooge, D.R.; Barner-Kowollik, C. Time-dependent differential and integral quantum yields for wavelength-dependent [4+4] photocycloadditions. Chem. Eur. J. 2020, 26, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Ermolaev, V.L. Ultrafast nonradiative transitions between higher excited states in organic molecules. Russ. Chem. Rev. 2001, 70, 471–490. [Google Scholar] [CrossRef]
- Tseng, H.-W.; Shen, J.-Y.; Kuo, T.-Y.; Tu, T.-S.; Chen, Y.-A.; Demchenko, A.P.; Chou, P.-T. Excited-state intramolecular proton-transfer reaction demonstrating anti-Kasha behavior. Chem. Sci. 2016, 7, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Popik, V.V. Wavelength-Dependent Photochemistry of Diazo Meldrum’s Acid and Its Spirocyclic Isomer, Diazirino Meldrum’s Acid: Wolff Rearrangement versus Isomerization. J. Am. Chem. Soc. 2003, 125, 1456–1457. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Tomin, V.I.; Chou, P.-T. Breaking the Kasha Rule for More Efficient Photochemistry. Chem. Rev. 2017, 117, 13353–13381. [Google Scholar] [CrossRef] [PubMed]
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Cherry, W.; Farneth, W. The effect of wavelength on organic photoreactions in solution. Reactions from upper excited states. Chem. Rev. 1978, 78, 125–145. [Google Scholar] [CrossRef]
- Shi, L.; Xie, X.; Troisia, A. Rapid calculation of internal conversion and intersystem crossing rate for organic materials discovery. J. Chem. Phys. 2022, 157, 134106. [Google Scholar] [CrossRef]
- Jabłoński, A. Efficiency of Anti-Stokes Fluorescence in Dyes. Nature 1933, 131, 839–840. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2006; ISBN 0-7167-8759-8. [Google Scholar]
- Itoh, T. Fluorescence and phosphorescence from higher excited states of organic molecules. Chem. Rev. 2012, 112, 4541–4568. [Google Scholar] [CrossRef]
- Del Valle, J.C.; Catalán, J. A Reappraisal of Kasha’s Rule. Phys. Chem. Chem. Phys. 2019, 21, 10061–10069. [Google Scholar] [CrossRef] [PubMed]
- Vavilov, S.I. The dependence of the intensity of the fluorescence of dyes upon the wave-length of the exciting light. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1922, 43, 307–320. [Google Scholar] [CrossRef]
- Braslavsky, S.E. Glossary of terms used in photochemistry (IUPAC). Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar] [CrossRef]
- Lewis, G.N.; Kasha, M. Phosphorescence and the Triplet State. J. Am. Chem. Soc. 1944, 66, 2100–2116. [Google Scholar] [CrossRef]
- Klan, P.; Wirz, J. Photochemistry of Organic Compounds: From Concepts to Practice; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Pianowski, Z.L. (Ed.) Molecular Photoswitches: Chemistry, Properties, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2022; ISBN 978-3-527-35104-6. [Google Scholar]
- Wayne, R.P. Principles and Applications of Photochemistry; Oxford University Press: Cary, NC, USA, 1988; ISBN 9780198552345/0198552343. [Google Scholar]
- Rosspeintner, A.; Lang, B.; Vauthey, E. Ultrafast Photochemistry in Liquids. Annu. Rev. Phys. Chem. 2013, 64, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, T.; Lang, B.; Rosspeintner, A.; Vauthey, E. Ultrafast elementary photochemical processes of organic molecules in liquid solution. Chem. Rev. 2017, 117, 10826–10939. [Google Scholar] [CrossRef] [PubMed]
- Livshits, M.Y.; Wang, L.; Vittardi, S.B.; Ruetzel, S.; King, A.; Brixner, T.; Rack, J.J. An excited state dynamics driven reaction: Wavelength-dependent photoisomerization quantum yields in [Ru(bpy)2(dmso)2]2+. Chem. Sci. 2020, 11, 5797–5807. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, USA, 2010; ISBN 1-891389-25-4. [Google Scholar]
- Yu, J.; Ma, H.; Huang, W.; Liang, Z.; Zhou, K.; Lv, A.; Li, X.-G.; He, Z. Purely Organic Room-Temperature Phosphorescence Endowing Fast Intersystem Crossing from Through-Space Spin−Orbit Coupling. J. Am. Chem. Soc. Au. 2021, 1, 1694–1699. [Google Scholar] [CrossRef]
- Levine, S.Z.; Knight, A.R.; Steer, R.P. Fluorescence from the Second Excited Singlet State of Thiophosgene Vapour. Chem. Phys. Lett. 1974, 29, 73–76. [Google Scholar] [CrossRef]
- Oka, T.; Knight, A.R.; Steer, R.P. A spectroscopic study of fluorescence from the second excited singlet state of thiophosgene vapor. J. Chem. Phys. 1975, 63, 2414–2420. [Google Scholar] [CrossRef]
- Steer, R.P. Structure and decay dynamics of electronic excited states of thiocarbonyl compounds. Rev. Chem. Intermed. 1981, 4, 1–41. [Google Scholar] [CrossRef]
- Acuña, A.U.; Catalán, J.; Toribio, F. Photon energy relaxation and thermal effects on gas-phase electronically excited methyl salicylate. J. Phys. Chem. 1981, 85, 241–245. [Google Scholar] [CrossRef]
- Catalán, J.; Del Valle, J.C.; Palomar, J.; Díaz, C.; De Paz, J.L.G. The six-membered intramolecular hydrogen bond position as a switch for inducing an excited state intramolecular proton transfer (ESIPT) in esters of o-Hydroxynaphthoic Acids. J. Phys. Chem. A 1999, 50, 10921–10934. [Google Scholar] [CrossRef]
- Viswanath, G.; Kasha, M. Confirmation of the Anomalous Fluorescence of Azulene. J. Chem. Phys. 1956, 24, 574–577. [Google Scholar] [CrossRef]
- Binsch, G.; Heilbronner, E.; Jankow, R.; Schmidt, D. On the fluorescence anomaly of azulene. Chem. Phys. Lett. 1967, 1, 135–138. [Google Scholar] [CrossRef]
- Birks, J.B. The photophysics of azulene. Chem. Phys. Lett. 1972, 17, 370–372. [Google Scholar] [CrossRef]
- Prlj, A.; Begušić, T.; Zhang, Z.T.; Fish, G.C.; Wehrle, M.; Zimmermann, T.; Choi, S.; Roulet, J.; Moser, J.-E.; Vaníček, J. Semiclassical approach to photophysics beyond Kasha’s rule and vibronic spectroscopy beyond the condon approximation. The case of azulene. J. Chem. Theory Comput. 2020, 16, 2617–2626. [Google Scholar] [CrossRef] [PubMed]
- Geldof, P.A.; Rettschnick, R.P.H.; Hoytink, G.J. Fluorescence from the second excited singlets of pyrene and 3,4-benzpyrene. Chem. Phys. Lett. 1969, 4, 59–61. [Google Scholar] [CrossRef]
- Peterson, D.L.; Lytle, F.E.; Laurendeau, N.M. Determination of flame temperature using the anomalous fluorescence of pyrene. Opt. Lett. 1986, 11, 345–347. [Google Scholar] [CrossRef]
- Easterly, C.E.; Christophorou, L.G.; Carter, J.G. Fluorescence from the second excited π-singlet state of aromatic hydrocarbons in solution. J. Chem. Soc. Faraday Trans. 1973, 69, 471–483. [Google Scholar] [CrossRef]
- Nakajima, A.; Baba, H. Fluorescence spectrum of pyrene vapor: Emission from the second excited singlet state. Bull. Chem. Soc. Jpn. 1970, 43, 967. [Google Scholar] [CrossRef]
- Baba, H.; Nakajima, A.; Aoi, M.; Chihara, K.J. Fluorescence from the second excited singlet state and radiationless processes in pyrene vapor. J. Chem. Phys. 1971, 55, 2433–2438. [Google Scholar] [CrossRef]
- Choi, I.S.; Lee, S.K. Analysis of Rotationally Cooled Vibronic Emission Spectra () of m-Xylyl Radical. Bull. Korean Chem. Soc. 1996, 17, 749–753. [Google Scholar]
- Uttamlal, M.; Holmes-Smith, A.S. The excitation wavelength dependent fluorescence of porphyrins. Chem. Phys. Lett. 2008, 454, 223–228. [Google Scholar] [CrossRef]
- Malpicci, D.; Lucenti, E.; Giannini, C.; Forni, A.; Botta, C.; Cariati, E. Prompt and long-lived anti-Kasha emission from organic dyes. Molecules 2021, 26, 6999. [Google Scholar] [CrossRef] [PubMed]
- Pavlovich, V.S. Dependence of excitation spectra of solutions of dipolar molecules on registration wavelength. Zh. Prikl. Spektr. 1976, 25, 480–487. [Google Scholar]
- Cushing, S.K.; Li, M.; Huang, F.; Wu, N. Origin of strong excitation wavelength dependent fluorescence of graphene oxide. ACS Nano 2014, 8, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. The red-edge effects: 30 years of exploration. Luminescence 2002, 17, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A. Dynamic Stokes shift and excitation wavelength dependent fluorescence of dipolar molecules in room temperature ionic liquids. J. Phys. Chem. B 2006, 110, 13704–13716. [Google Scholar] [CrossRef]
- Khara, D.C.; Samanta, A. Solvation dynamics and red-edge effect of two electrically charged solutes in an imidazolium ionic liquid. Indian. J. Chem. 2010, 49, 714–720. [Google Scholar]
- Józefowicz, M.; Heldt, J.R. Excitation-wavelength dependent fluorescence of ethyl 5-(4-aminophenyl)-3-amino2,4-dicyanobenzoate. J. Fluoresc. 2011, 21, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Galley, W.C.; Purkey, R.M. Role of heterogeneity of the solvation site in electronic spectra in solution. Proc. Natl. Acad. Sci. USA 1970, 67, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Milton, J.C.; Purkey, R.M.; Galley, W.C. The kinetics of solvent reorientation in hydroxylated solvents from the exciting-wavelength dependence of chromophore emission spectra. J. Chem. Phys. 1978, 68, 5396–5404. [Google Scholar] [CrossRef]
- Pavlovich, V.S.; Pershukevich, P.P.; Pikulik, L.G. Instantanous phosphorescence spectra of glassy polar solutions of acetyl phthalamide derivatives. Zh. Prikl. Spektr. 1983, 39, 779–785. [Google Scholar]
- Khisamov, R.M.; Ryadun, A.A.; Sukhikh, T.S.; Konchenko, S.N. Excitation wavelength-dependent room-temperature phosphorescence: Unusual properties of novel phosphinoamines. Mol. Syst. Des. Eng. 2021, 6, 1056–1065. [Google Scholar] [CrossRef]
- Xiao, G.; Ma, Y.-J.; Qi, Z.; Fang Chena, T.; Yan, D. A flexible ligand and halogen engineering enable one phosphor-based full-colour persistent luminescence in hybrid perovskitoids. Chem. Sci. 2024, 15, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Holovka, J.M.; Gardner, P.D. Photolysis and photoisomerization of the benzene oxide-oxepin system. J. Am. Chem. Soc. 1967, 89, 6390–6391. [Google Scholar] [CrossRef]
- Dauben, W.G.; Kellog, M.S. Wavelength dependency in the photochemistry of 1,3-cyclohexadienes. cis-Bicyclo [4.3.0]nona-2,4-diene to cis,cis,trans-1,3,5-cyclononatriene valence tautomerism. J. Am. Chem. Soc. 1971, 93, 3805–3807. [Google Scholar] [CrossRef]
- Zimmerman, G.; Chow, L.-Y.; Paik, U.-J. The photochemical isomerization of azobenzene. J. Am. Chem. Soc. 1958, 80, 3528–3531. [Google Scholar] [CrossRef]
- Rau, H. Azo Compounds. In Advances in Photochemistry; Neckers, D.C., Volman, D.H., von Bunau, G., Eds.; Wliley & Sons: New York, NY, USA, 1995; Volume 19, ISBN 0-471-04912-3. [Google Scholar]
- Montali, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; Taylor and Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Rau, H. Photochromism: Molecules and Systems; Durr, H., Bounas-Laurent, H., Eds.; Elsevier: Amsterdam, The Netherland, 2003; Volume 1, ISBN 9780444513229. [Google Scholar]
- Gagliardi, L.; Orlandi, G.; Bernardi, F.; Cembran, A.; Garavelli, M. A theoretical study of the lowest electronic states of azobenzene: The role of torsion coordinate in the cis-trans photoisomerization. Theor. Chem. Acc. 2004, 111, 363–372. [Google Scholar] [CrossRef]
- Ladányi, V.; Dvořák, P.; Al-Anshori, J.; Vetráková, L.; Wirz, J.; Heger, D. Azobenzene photoisomerization quantum yields in methanol redetermined. Photochem. Photobiol. Sci. 2017, 16, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Mauser, H.; Gauglitz, G.; Compton, R.G.; Hancock, G. (Eds.) Comprehensive Chemical Kinetics, Photokinetics: Theoretical Fundamentals and Applications; Elsevier: Amesterdam, The Netherland, 1998; Volume 36, ISBN 978-0444825360. [Google Scholar]
- Stranius, K.; Börjesson, K. Determining the Photoisomerization Quantum Yield of Photoswitchable Molecules in Solution and in the Solid State. Sci. Rep. 2017, 7, 41145. [Google Scholar] [CrossRef] [PubMed]
- Gade, R.; Porada, T. Determination of quantum yields and product spectra by studying reversible photoisomerizations in solution. J. Photochem. Photobiol. A Chem. 1997, 107, 27–34. [Google Scholar] [CrossRef]
- Deniel, M.H.; Lavabre, D.; Micheau, J.C. Photokinetics under continuous irradiation. In Organic Photochromic and Thermochromic Compounds; Crano, J.C., Guglielmetti, R.J., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; ISBN 978-0-306-45882-8. [Google Scholar] [CrossRef]
- Delbaere, S.; Vermeersh, G.; Micheau, J.C. Quantitative analysis of the dynamic behaviour of photochromic systems. J. Photochem. Photobiol. C Chem. Photochem. Rev. 2011, 12, 74–105. [Google Scholar] [CrossRef]
- Lewis, G.N.; Milgel, T.T.; Lipkin, D. The Absorption and Re-emission of Light by cis- and trans-Stilbenes and the Efficiency of their Photochemical Isomerization. J. Am. Chem. Soc. 1940, 62, 2973–2980. [Google Scholar] [CrossRef]
- Olson, A.R. The study of chemical reactions from potential energy diagrams. Trans. Faraday Soc. 1931, 27, 69–76. [Google Scholar] [CrossRef]
- Hartley, G.S. The cis-form of azobenzene and the velocity of the thermal cis→trans-conversion of azobenzene and some derivatives. J. Chem. Soc. 1938, 633–642. [Google Scholar] [CrossRef]
- Dhammika Bandara, H.M.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Diau, E.W.-G. A new trans-to-cis photoisomerization mechanism of azobenzene on the S1(n, π*) surface. J. Phys. Chem. A 2004, 108, 950–956. [Google Scholar] [CrossRef]
- Samai, S.; Bradley, D.J.; Choi, T.L.Y.; Yan, Y.; Ginger, D.S. Temperature dependent photoisomerization quantum yield for azobenzene-modified DNA. J. Phys. Chem. C 2017, 121, 6997–7004. [Google Scholar] [CrossRef]
- Shi, Y.-B.; Hearst, J.E. Wavelength dependence for the photoreactions of DNA-psoralen monoadducts. 2: Photo-cross-linking of monoadducts. Biochemistry 1987, 26, 3792–9798. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.E.; Goodeve, C.F.; Lythgoe, R.J. The spectral variation of the photosensitivity of visual purple. Proc. Natl. Acad. Sci. USA 1939, 170, 102–112. [Google Scholar]
- Kim, J.E.; Tauber, M.J.; Mathies, R.A. Wavelength-dependent cis-trans isomerization in vision. Biochemistry 2001, 40, 13774–13778. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Tauber, M.J.; Mathies, R.A. Analysis of the mode specific excited-state energy distribution and wavelength-dependent photoreaction quantum yield in rhodopsin. Biophys. J. 2003, 84, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Stock, G. Quantum mechanical modelling of the femtosecond isomerization of rhodopsin. J. Phys. Chem. 2000, 104, 1146–1149. [Google Scholar] [CrossRef]
- Tscherbul, T.V.; Brumer, P. Escitation of biomolecules with incoherent light: Quantum yield for the photoisomerization of model retinal. J. Phys. Chem. 2014, 118, 3100–3111. [Google Scholar] [CrossRef]
- Balke, D.E.; Becker, R.S. Relationship between the absorption and excitation spectra and relative quantum yields of fluorescence of all-trans-retinal. J. Am. Chem. Soc. 1968, 90, 6710–6711. [Google Scholar] [CrossRef]
- Dartnall, H.J.A. The photosensitivities of visual pigments in the presence of hydroxylamine. Vision. Res. 1968, 8, 339–358. [Google Scholar] [CrossRef]
- Stensitzki, T.; Muders, V.; Schlesinger, R.; Heberle, J.; Heyne, K. The primary photoreaction of channelrhodopsin-1: Wavelength dependent photoreactions induced by ground-state heterogeneity. Front. Mol. Biosci. 2015, 2, 41. [Google Scholar] [CrossRef]
- Schulte-Frohlinde, D.; Blume, H.; Gusten, H. Photochemical cis-trans-isomerization of substituted stilbene. J. Phys. Chem. 1962, 66, 2486–2491. [Google Scholar] [CrossRef]
- Gorner, H.; Kuhn, H.J. Cis-trans isomerization of stilbenes and stilbene-like molecules. In Advances in Photochemistry; Neckers, D.C., Volman, D.H., von Bunau, G., Eds.; Wliley & Sons: New York, NY, USA, 1995; Volume 19, ISBN 0-471-04912-3. [Google Scholar]
- Maafi, M.; Maafi, W. Φ-Order kinetics of photoreversible-drug reactions. Int. J. Pharm. 2014, 471, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Maafi, W. Quantitative assessment of photostability and photostabilisation of fluvoxamine and its design for actinometry. Photochem. Photobiol. Sci. 2015, 14, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Lee, L.Y. Actinometric and Φ-order photodegradation of anti-cancer Sunitinib. J. Pharm. Biomed. Anal. 2015, 110, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Maafi, W. Montelukast photodegradation: Elucidation of Φ-order kinetics, determination of quantum yields and application to actinometry. Int. J. Pharm. 2014, 471, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Al-Qarni, M.A. Φ-order spectrophotokinetic characterisation and quantification of trans-cis oxyresveratrol reactivity, photodegradation and actinometry. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 188, 64–71. [Google Scholar] [CrossRef]
- Maafi, M.; Al-Qarni, M.A. Dependence of pinosylvin photochemical quantum yield on excitation wavelength. Trends Photochem. Photobiol. 2019, 18, 45–58. [Google Scholar]
- Maafi, M. On photokinetics under monochromatic light. Front. Chem. 2023, 11, 1233151. [Google Scholar] [CrossRef]
- Maafi, M. On photokinetics under polychromatic light. Front. Chem. 2024, 12, 1367276. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.; Moer, J.F. Photochemische Reaktionen. 50. Mitteilung [1]. Zur Photochemie von gesättigten β-Ketosulfiden II. Synthese von 6-Hydroxy-2-thia-7-oxa-isotwistan. Helv. Chim. Acta 1969, 52, 725–737. [Google Scholar] [CrossRef]
- Pacifici, J.G.; Diebert, C. Wavelength effect in photolysis of 4,4,6,6-tetramethyl-1-thiaspiro [2.3]hexan-5-one. J. Am. Chem. Soc. 1969, 91, 4595. [Google Scholar] [CrossRef]
- Gisin, M.; Wirz, J. Photolysis of the azo-precursors of 2,3- and 1,s-naphtoquinodimethane. Helv. Chim. Acta 1976, 59, 2273–2277. [Google Scholar] [CrossRef]
- Pagni, R.M.; Burnett, M.N.; Dodd, J.R. Photochemistry of 1,4-Dihydronaphtho [1,8-de] [1,2] diazepine. Preparation and electron spin resonance observation of the uinsubstituted 1,8-Naphthoquinodimethane. J. Am. Chem. Soc. 1977, 99, 1972–1973. [Google Scholar] [CrossRef]
- Bochet, C.G. Chromic orthogonality in organic synthesis. Synlett 2004, 13, 2268–2274. [Google Scholar] [CrossRef]
- Bochet, C.G. Photochemical release of various functional groups. Pure Appl. Chem. 2006, 78, 241–247. [Google Scholar] [CrossRef]
- Protti, S.; Ravelli, D.; Fagnoni, M. Wavelength-dependence and wavelength-selectivity in photochemical reactions. Photochem. Photobiol. Sci. 2019, 18, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Bochet, C.G. Wavelength-selective cleavage of photolabile protecting groups. Tetrahedron Lett. 2000, 41, 6341–6346. [Google Scholar] [CrossRef]
- Bochet, C.G. Orthogonal photolysis of protecting Groups. Angew. Int. Ed. 2001, 40, 2071–2073. [Google Scholar] [CrossRef]
- Stegmaier, P.; Alonso, J.M.; del Campo, A. Photoresponsive Surfaces with Two Independent Wavelength-Selective Functional Levels. Langmuir 2008, 24, 11872–11879. [Google Scholar] [CrossRef] [PubMed]
- Kotzur, N.; Briand, B.; Beyermann, M.; Hagen, V. Wavelength-selective photoactivatable protecting groups for thiols. J. Am. Chem. Soc. 2009, 25, 16927–16931. [Google Scholar] [CrossRef]
- Kantevari, S.; Matsuzaki, M.; Kanemoto, Y.; Kasai, H.; Ellis-Davies, G.C.R. Two-color, two-photon uncaging of glutamate and GABA. Nat. Methods 2010, 7, 123–125. [Google Scholar] [CrossRef]
- San Miguel, V.; Bochet, C.G.; del Campo, A. Wavelength-selective caged surfaces: How many functional levels are possible? J. Am. Chem. Soc. 2011, 133, 5380–5388. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.; Joshi, K.B.; Fichte, M.A.H.; Mack, T.; Wachtveitl, J.; Heckel, A. Wavelength-selective uncaging of dA and dC residues. Org. Lett. 2011, 13, 1450–1453. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; San Miguel, V.; del Campo, A. Light-triggered multifunctionality at surfaces mediated by photolabile protecting groups. Macromol. Rapid. Commun. 2013, 34, 310–329. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernández, L.; Herbivo, C.; San Miguel Arranz, V.; Warther, D.; Donato, L.; Specht, A.; del Campo, A. Dual photosensitive polymers with wavelength-selective photoresponse. Adv. Mater. 2014, 26, 5012–5017. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.J.; Velema, W.A.; Lerch, M.M.; Szymanski, W.; Feringa, B.L. Wavelength-selective cleavage of photoprotecting groups: Strategies and applications in dynamic systems. Chem. Soc. Rev. 2015, 44, 3358–3377. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, N.; Ciftci, M.; Jung, K.; Boyer, C. Mediating Reaction Orthogonality in Polymer and Materials Science. Angew. Chem. Int. Ed. 2021, 60, 1748–1781. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, J.; Sinsel, F.; Heckel, A. Chromatic Selectivity of Coumarin-Caged Oligonucleotides. Chem. Eur. J. 2023, 29, e202204014. [Google Scholar] [CrossRef] [PubMed]
- Dolinski, N.D.; Page, Z.A.; Callaway, E.B.; Eisenreich, F.; Garcia, R.V.; Chavez, R.; Bothman, D.P.; Hecht, S.; Zok, F.W.; Hawker, C.J. Solution mask liquid lithography (SMaLL) for one-step, multimaterial 3D printing. Adv. Mater. 2018, 30, 1800364. [Google Scholar] [CrossRef] [PubMed]
- Menzel, J.P.; Feist, F.; Tuten, B.; Weil, T.; Blinco, J.P.; Barner-Kowollik, C. Light-Controlled Orthogonal Covalent Bond Formation at Two Different Wavelengths. Angew. Chem. Int. Ed. 2019, 58, 7470–7474. [Google Scholar] [CrossRef]
- Birks, J.B. (Ed.) Organic Molecular Photophysics; Wiley-Interscience: New York, NY, USA, 1973; Volume I, ISBN 10.0471074152. [Google Scholar]
- Nobs, F.; Burger, U.; Schaffner, K. Specifically (π → π*)-Induced Cyclohexenone Reactions: 4a-(Z-1-Propenyl)-bicyclo [4.4.0]dec-1(8a)-en-2-one and 4a-Z-1-Propenyl-bicyclo [4.4.0]deca-1(8a), 7-dien-2-one. Helv. Chim. Acta 1977, 60, 1607–1628. [Google Scholar] [CrossRef]
- Adriano, N.; Ahearn, C.; Black, C.; Cracchiolo, M.; Ghere, D.; Nuñez, A.; Olivan, L.; Patel, R.; Saner, S.; Smith, K.R.; et al. Solvent- and Wavelength-Dependent Photolysis of Estrone. Photochem. Photobiol. 2022, 98, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Bridge, N.K. Effect of wavelength on the flash photolysis of quinones. Trans. Faraday Soc. 1960, 56, 1001–1007. [Google Scholar] [CrossRef]
- Gloor, J.; Schaffner, K. Spezifisch π→π*-induzierte Reaktionen von γ-Dimethoxymethylcyclo hexen-2-onen: 1,3-Umlagerung und Wasserstoffabstraktion durch das α-Kohlenstoffatom. Helv. Chim. Acta 1974, 57, 1815–1845. [Google Scholar] [CrossRef]
- Dunkel, P. Photoremovable Protecting Groups. Encyclopedia 2022, 2, 1225–1236. [Google Scholar] [CrossRef]
- Saracini, C.; Liakos, D.G.; Zapata Rivera, J.E.; Neese, F.; Meyer, G.J.; Karlin, K.D. Excitation wavelength dependent O2 release from copper(II)-superoxide compounds: Laser flash-photolysis experiments and theoretical studies. J. Am. Chem. Soc. 2014, 136, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Ziani, Z.; Loiseau, F.; Lognon, E.; Boggio-Pasqua, M.; Philouze, C.; Cobo, S.; Royal, G.C. Synthesis of a negative photochrome with high switching quantum yields and capable of singlet-oxygen production and storage. Chem. Eur. J. 2021, 27, 16642–16653. [Google Scholar] [CrossRef] [PubMed]
- Thoma, K.; Klimek, R. Photostabilisation of drugs in dosage forms without protection from packaging materials. Int. J. Pharm. 1991, 67, 169–175. [Google Scholar] [CrossRef]
- Maafi, W.; Maafi, M. Modelling nifedipine photodegradation, photostability and actinometry properties. Int. J. Pharm. 2013, 456, 153–164. [Google Scholar] [CrossRef]
- Maafi, M.; Maafi, W. Quantification of Unimolecular Photoreaction Kinetics: Determination of Quantum Yields and Development of Actinometers-The Photodegradation Case of Cardiovascular Drug Nisoldipine. Int. J. Photoenergy 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Maafi, M.; Lee, L.Y. Determination of dacarbazine Φ-order photokinetics, quantum yields, and potential for actinometry. J. Pharm. Sci. 2015, 104, 3501–3509. [Google Scholar] [CrossRef]
- El-Hendawy, M.M.; Fayed, T.A.; Awad, M.K.; English, N.J.; Etaiw, S.E.H.; Zaki, A.B. Photophysics, photochemistry and thermal stability of diarylethene-containing benzothiazolium species. J. Photochem. Photochem. Chem. A 2015, 301, 20–31. [Google Scholar] [CrossRef]
- Ribeiro, A.; Ballardini, R.; Belser, P.; Gandolfi, M.T.; Iyer, V.M.; Moggi, L. Photochemical investigation of photochromic diaryethene compound that can be used as a wide range actinometer. Photochem. Photobiol. Sci. 2009, 8, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Ern, J.; Bens, A.T.; Martin, H.-D.; Kuldova, K.; Trommsdorff, H.P.; Kryschi, C. Ring-opening and -closure reaction dynamics of a photochromic dithienylethene derivative. J. Phys. Chem. A 2002, 106, 1654–1660. [Google Scholar] [CrossRef]
- Herder, M.; Schmidt, B.M.; Grubert, L.; Patzel, M.; Schwarz, J.; Hecht, S. Improving the Fatigue Resistance of Diarylethene Switches. J. Am. Chem. Soc. 2015, 137, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Maafi, M.; Brown, R.G. Kinetic analysis and elucidation options for AB(1k,2F) systems. New spectrokinetic methods for photochromes. Photochem. Photobiol. Sci. 2008, 7, 1360–1372. [Google Scholar] [CrossRef]
- Maafi, M.; Brown, R.G. The kinetic model for AB(1F) systems. A closed-form integration of the differential equation with a variable photokinetic factor. J. Photochem. Photobiol. A Chem. 2007, 187, 319–324. [Google Scholar] [CrossRef]
- Maafi, M.; Al-Qarni, M.A. Mono- and polychromatic light diarylethene-actinometer for the visible range. Dye. Pigment. 2022, 198, 109942. [Google Scholar] [CrossRef]
- Sumi, T.; Takagi, Y.; Yagi, A.; Morimoto, M.; Irie, M. Photoirradiation wavelength dependence of cycloreversion quantum yields of diarylethenes. Chem. Commun. 2014, 50, 3928–3930. [Google Scholar] [CrossRef]
- Cipolloni, M.; Gentili, P.L.; Ortica, F.; Becker, R.S.; Favaro, G. Effects of solvent, excitation wavelength, and concentration on the photobehavior of some diazonaphthoquinones. Arkivoc 2011, 9, 205–220. [Google Scholar] [CrossRef]
- Di Nunzio, M.R.; Gentili, P.L.; Romani, A.; Favaro, G. Photochromic, thermochromic, and fluorescent spirooxazines and naphthopyrans: A spectrokinetic and thermodynamic study. Chemphyschem 2008, 9, 768–775. [Google Scholar] [CrossRef]
- Wilkinson, F.; Hobley, J.; Naftaly, M. Photochromism of spiro-naphthoxazines: Molar absorption coefficients and quantum efficiencies. J. Chem. Soc. Faraday Trans. 1992, 88, 1511–1517. [Google Scholar] [CrossRef]
- Fischer, E. Calculation of photostationary states in systems A.dblarw. B when only A is known. J. Phys. Chem. 1967, 71, 3704–3706. [Google Scholar] [CrossRef]
- Kaiser, C.; Halbritter, T.; Heckel, A.; Wachtveitl, J. Thermal, Photochromic and Dynamic Properties of Water-Soluble Spiropyrans. ChemistrySelect 2017, 2, 4111–4123. [Google Scholar] [CrossRef]
- Ortica, F.; Smimmo, P.; Favaro, G.; Mazzucato, U.; Delbaere, S.; Venec, D.; Vermeersch, G.; Frigoli, M.; Moustrou, C.; Samat, A. Effect of oligothiophene substituents on the photophysical and photochemical properties of a naphthopyran. Photochem. Photobiol. Sci. 2004, 3, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Ortica, F.; Moustrou, C.; Berthet, J.; Favaro, G.; Samat, A.; Guglielmetti, R.; Vermeersch, G.; Mazzucato, U. Comprehensive photokinetic and NMR study of a biphotochromic supermolecule involving two naphthopyrans linked to a central thiophene unit through acetylenic bonds. Photochem. Photobiol. 2003, 78, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Tekahashi, Y. Trifluoromethyl-substituted photochromic indolylfulgide. A remarkably durable fulgide towards photochemical and thermal treatments. Chem. Lett. 1996, 25, 1037–1038. [Google Scholar] [CrossRef]
- Wolak, M.A.; Gillespie, N.B.; Thomas, C.J.; Birge, R.R.; Lees, W.J. Optical properties of photochromic fluorinated indolylfulgides. J. Photochem. Photobiol. A Chem. 2001, 144, 83–91. [Google Scholar] [CrossRef]
- Wolak, M.A.; Gillespie, N.B.; Thomas, C.J.; Birge, R.R.; Lees, W.J. Thermolysis of fluorinated cycloalkylidene fulgides yields a new class of photochromic compounds. Chem. Commun. 2003, 992–993. [Google Scholar] [CrossRef]
- Cordes, T.; Malkmus, S.; DiGirolamo, J.A.; Lees, W.J.; Nenov, A.; de Vivie-Riedle, R.; Braun, M.; Zinth, W. Accelerated and efficient photochemistry from higher excited electronic states in fulgide molecules. J. Phys. Chem. A 2008, 112, 13364–13371. [Google Scholar] [CrossRef]
- Reinfelds, M.; Hermanns, V.; Halbritter, T.; Wachtveitlz, J.; Braun, M.; Slanina, T.; Heckel, A. A robust, broad-absorbing fulgide derivative as a universal chemical actinometer for UV to NIR region. ChemPhotoChem 2019, 3, 441–449. [Google Scholar] [CrossRef]
- Volarić, J.; Szymanski, W.; Simeth, N.A.; Feringa, B.L. Molecular photoswitches in aqueous environments. Chem. Soc. Rev. 2021, 50, 12377–12449. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhang, J. (Eds.) Photochromic Materials: Preparation, Properties and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; ISBN 978-3-527-68372-7. [Google Scholar]
- Maafi, M.; Maafi, W. Modelling and elucidation of the kinetics of multiple consecutive photoreactions AB4(4Φ) with Φ-order kinetics. Application to the photodegradation of ribofavin. J. Pharm. Sci. 2016, 105, 3537–3548. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.S.; Dolan, E.; Balke, D.E. Vibronic effects in photochemistry. Competition between internal conversion and photochemistry. J. Chem. Phys. 1969, 50, 239–245. [Google Scholar] [CrossRef]
- Becker, R.S.; Michl, J. Photochromism of synthetic and naturally occurring 2H-chromenes and 2H-pyrans. J. Am. Chem. Soc. 1966, 88, 5931–5933. [Google Scholar] [CrossRef]
- Lenoble, C.; Becker, R.S. Photophysics, photochemistry and kinetics of photochromic 2H-pyrans and chromenes. J. Photochem. 1986, 33, 187–197. [Google Scholar] [CrossRef]
- Becker, R.S.; Favaro, G.; Romani, A.; Gentili, P.L.; Dias, F.M.B. Vibronic effects in pathways of photochemistry and vibrational relaxation. Chem. Phys. 2005, 316, 108–116. [Google Scholar] [CrossRef]
- Becker, R.S.; Pelliccioli, A.P.; Romani, A.; Favaro, G. Vibronic quantum effects in fluorescence and photochemistry. Competition between vibrational relaxation and photochemistry and consequences on photochemical control. J. Am. Chem. Soc. 1999, 121, 2104–2109. [Google Scholar] [CrossRef]
- Becker, R.S.; Favaro, G. New concepts in photochemistry and photophysics: Photochromic and other type molecules. J. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 167–176. [Google Scholar] [CrossRef]
- Zewail, A.H. Femtochemistry: Atomic-scale dynamics of the chemical bond using ultrafast lasers (Nobel Lecture). Angew. Chem. Int. Ed. 2000, 39, 2586–2631. [Google Scholar] [CrossRef]
- Tamai, N.; Miyasaka, H. Ultrafast dynamics of photochromic systems. Chem. Rev. 2000, 100, 1875–1890. [Google Scholar] [CrossRef]
- Briand, J.; Bram, O.; Rehault, J.; Leonard, J.; Cannizzo, A.; Chergui, M.; Zanirato, V.; Olivucci, M.; Helbing, J.; Haacke, S. Coherent ultrafast torsional motion and isomerization of a biomimetic dipolar photoswitch. Phys. Chem. Chem. Phys. 2010, 12, 3178–3187. [Google Scholar] [CrossRef] [PubMed]
- Wynne, K.; Hochstrasser, R.M. Coherence and adiabaticity in ultrafast electron transfer. Adv. Chem. Phys. Electron Transf. Isol. Mol. Biomol. Part 2 1999, 107, 263–309. [Google Scholar] [CrossRef]
- Rather, S.R.; Scholes, G.D. From fundamental theories to quantum coherences in electron transfer. J. Am. Chem. Soc. 2019, 141, 708–722. [Google Scholar] [CrossRef]
- Chudoba, C.; Riedle, E.; Pfeiffer, M.; Elsaesser, T. Vibrational Coherence in Ultrafast Excited State Proton Transfer. Chem. Phys. Lett. 1996, 263, 622–628. [Google Scholar] [CrossRef]
- Myahkostupov, M.; Pagba, C.V.; Gundlach, L.; Piotrowiak, P. Vibrational state dependence of interfacial electron transfer: Hot electron injection from the S1 state of azulene into TiO2 nanoparticles. J. Phys. Chem. C 2013, 117, 20485–20493. [Google Scholar] [CrossRef]
- Bishenden, E.; Donaldson, D.J. Mode-specific chemical branching ratios in the photodissociation of OClO. J. Chem. Phys. 1993, 99, 3129–3132. [Google Scholar] [CrossRef]
- Fidder, H.; Tschirschwitz, F.; Dühr, O.; Nibbering, E.T.J. Vibrational mode-specific photochemical reaction dynamics of chlorine dioxide in solution. J. Chem. Phys. 2001, 114, 6781–6794. [Google Scholar] [CrossRef]
- Wallace, P.; Bollinger, C.; Hayes, S.; Reid, A. On the actinic wavelength dependence of OClO photochemistry in solution. J. Chem. Phys. 2003, 116, 1883–1890. [Google Scholar] [CrossRef]
- Vaida, V.; Feierabend, K.J.; Rontu, N.; Takahashi, K. Sunlight-initiated photochemistry: Excited vibrational states of atmospheric chromophores. Int. J. Photoenergy 2008, 138091. [Google Scholar] [CrossRef]
- Reynard, L.M.; Donaldson, D.J. Overtone-induced chemistry of trifluoroacetic acid: An experimental and theoretical study. J. Phys. Chem. A 2002, 106, 8651–8657. [Google Scholar] [CrossRef]
- Staikova, M.; Oh, M.; Donaldson, D.J. Overtone-induced decarboxylation: A potential sink for atmospheric diacids. J. Phys. Chem. A 2005, 109, 597–602. [Google Scholar] [CrossRef]
- Young, C.J.; Donaldson, D.J. Overtone-induced degradation of perfluorinated alcohols in the atmosphere. J. Phys. Chem. A 2007, 111, 13466–13471. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, C.E.; Delgado, Y.P.; Sunuwar, S. Phase shift cavity ring down and FT-NIR spectra of CH vibrational transitions and band strengths of ethyl acetate. J. Quant. Spectr. Rad. Transf. 2024, 312, 108810. [Google Scholar] [CrossRef]
- Donaldson, D.J.; Tuck, A.F.; Vaida, V. Atmospheric photochemistry via vibrational overtone absorption. Chem. Rev. 2003, 103, 4717–4729. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Chaban, G.M.; Finlayson-Pitts, B.J.; Gerber, R.B. Photochemical processes induced by vibrational overtone excitations: Dynamics simulations for cis-HONO, trans-HONO, HNO3, and HNO3-H2O. J. Phys. Chem. A 2006, 110, 5342–5354. [Google Scholar] [CrossRef]
- Miller, Y.; Gerber, R.B. Dynamics of vibrational overtone excitations of H2SO4, H2SO4-H2O: Hydrogen-hopping and photodissociation processes. J. Am. Chem. Soc. 2006, 128, 9594–9595. [Google Scholar] [CrossRef] [PubMed]
- Goss, L.M.; Vaida, V.; Brault, J.W.; Skodje, R.T. Sequential two-photon dissociation of atmospheric water. J. Phys. Chem. A 2001, 105, 70–75. [Google Scholar] [CrossRef]
- Brown, S.S.; Metz, R.B.; Berghout, H.L.; Crim, F.F. Vibrationally mediated photodissociation of isocyanic acid (HNCO): Preferential N–H bond fission by excitation of the reaction coordinate. J. Chem. Phys. 1996, 105, 6293–9303. [Google Scholar] [CrossRef]
- Lai, H.Y.; Jhang, W.R.; Tseng, C.M. Mode-dependent excited-state lifetime of phenol under the S1/S2 conical intersection. J. Chem. Phys. 2018, 149, 031104. [Google Scholar] [CrossRef]
- Troe, J. Analysis of Quantum Yields for the Photolysis of Formaldehyde at λ > 310 nm. J. Phys. Chem. A 2007, 111, 3868–3874. [Google Scholar] [CrossRef]
- Röth, E.P.; Ehhalt, D.H. A simple formulation of the CH2O photolysis quantum yields. Atmos. Chem. Phys. 2015, 15, 7195–7202. [Google Scholar] [CrossRef]
- Jankowski, J.J.; Kieber, D.J.; Mopper, K.; Neale, P.J. Development and intercalibration of ultraviolet solar actinometer. Photochem. Photobiol. 2000, 71, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Hatchard, C.G.; Parker, C.A. A New Sensitive Chemical Actinometer. II. Potassium Ferrioxalate as a Standard Chemical Actinometer. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1956, 235, 518–536. [Google Scholar] [CrossRef]
- Favaro, G.; di Nunzio, M.R.; Gentili, P.L.; Romani, A.; Becker, R.S. Effects of the Exciting Wavelength and Viscosity on the Photobehavior of 9- and 9,10-Bromoanthracenes. J. Phys. Chem. A 2007, 111, 5948–5953. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Silva, E.; Reis, R.L. Light-triggered release of photocaged therapeutics—Where are we now? J. Contr. Release 2019, 298, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B. The wavelengths in sunlight effective in producing skin cancer: A theoretical analysis. Proc. Natl. Acad. Sci. USA 1974, 71, 3363–3366. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible light-induced photocatalytic reaction of gold-modified titanium(iv) oxide particles: Action spectrum analysis. Chem. Commun. 2009, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-K.; Mills, A.; O’rourke, C. Action spectra in semiconductor photocatalysis. Chem. Soc. Rev. 2017, 46, 4877–4894. [Google Scholar] [CrossRef] [PubMed]
- Datcu, A.; Mendoza, M.L.; Pérez del Pino, A.; Logofatu, C.; Luculescu, C.; György, E. UV–vis light induced photocatalytic activity of TiO2/graphene oxide nanocomposite coatings. Catal. Today 2019, 321–322, 81–86. [Google Scholar] [CrossRef]
- Pettersson, L.A.A.; Roman, L.S.; Inganäs, O. Modeling photocurrent action spectra of photovoltaic devices based on organic thin films. J. Appl. Phys. 1999, 86, 487–496. [Google Scholar] [CrossRef]
- Langford, C.H.; Achari GIzadifard, M. Wavelength dependence of luminescence and quantum yield in dichlorination of PCBs. J. Photochem. Photobiol. A Chem. 2011, 222, 40–46. [Google Scholar] [CrossRef]
- Bhat, A.P.; Pomerantz, W.C.K.; Arnold, W.A. Wavelength-Dependent UV-LED Photolysis of Fluorinated Pesticides and Pharmaceuticals. Environ. Sci. Technol. 2023, 57, 5327–5336. [Google Scholar] [CrossRef] [PubMed]
- Kuldova, K.; Tsyganenko, K.; Corval, A.; Trommsdorff, H.P.; Bens, A.T.; Kryschi, C. Photo-switchable dithienylethenes: Threshold of the photoreactivity. Synth. Met. 2000, 115, 163–166. [Google Scholar] [CrossRef]
- Bens, A.T.; Frewert, D.; Kodatis, K.; Kryschi, C.; Martin, H.-D.; Trommsdorff, H.P. Coupling of Chromophores: Carotenoids and Photoactive Diarylethenes–Photoreactivity versus Radiationless Deactivation. Eur. J. Org. Chem. 1998, 11, 2333–2338. [Google Scholar] [CrossRef]
- Irie, M.; Eriguchi, T.; Takada, T.; Uchida, K. Photochromism of diarylethenes having thiophene oligomers as the aryl groups. Tetrahedron 1997, 53, 12263–12271. [Google Scholar] [CrossRef]
- Nagorny, S.; Schewe, M.; Weingartz, T.; Eitzeroth, A.; Adams, J.; Rembe, C.; Schmidt, A. Stabilities of bis(thienyl)ethenes in polymethyl methacrylate (PMMA) coatings as absorbance modulation layers for nanoscale imaging. Mater. Adv. 2024, 5, 159–170. [Google Scholar] [CrossRef]
- Maafi, M. Useful Spectrokinetic Methods for the Investigation of Photochromic and Thermo-Photochromic Spiropyrans. Molecules 2008, 13, 2260–2302. [Google Scholar] [CrossRef]
- Dulić, D.; Kudernac, T.; Pužys, A.; Feringa, B.L.; van Wees, B.J. Temperature gating of the ring-opening process in diarylethene molecular switches. Adv. Mater. 2007, 19, 2898–2902. [Google Scholar] [CrossRef]
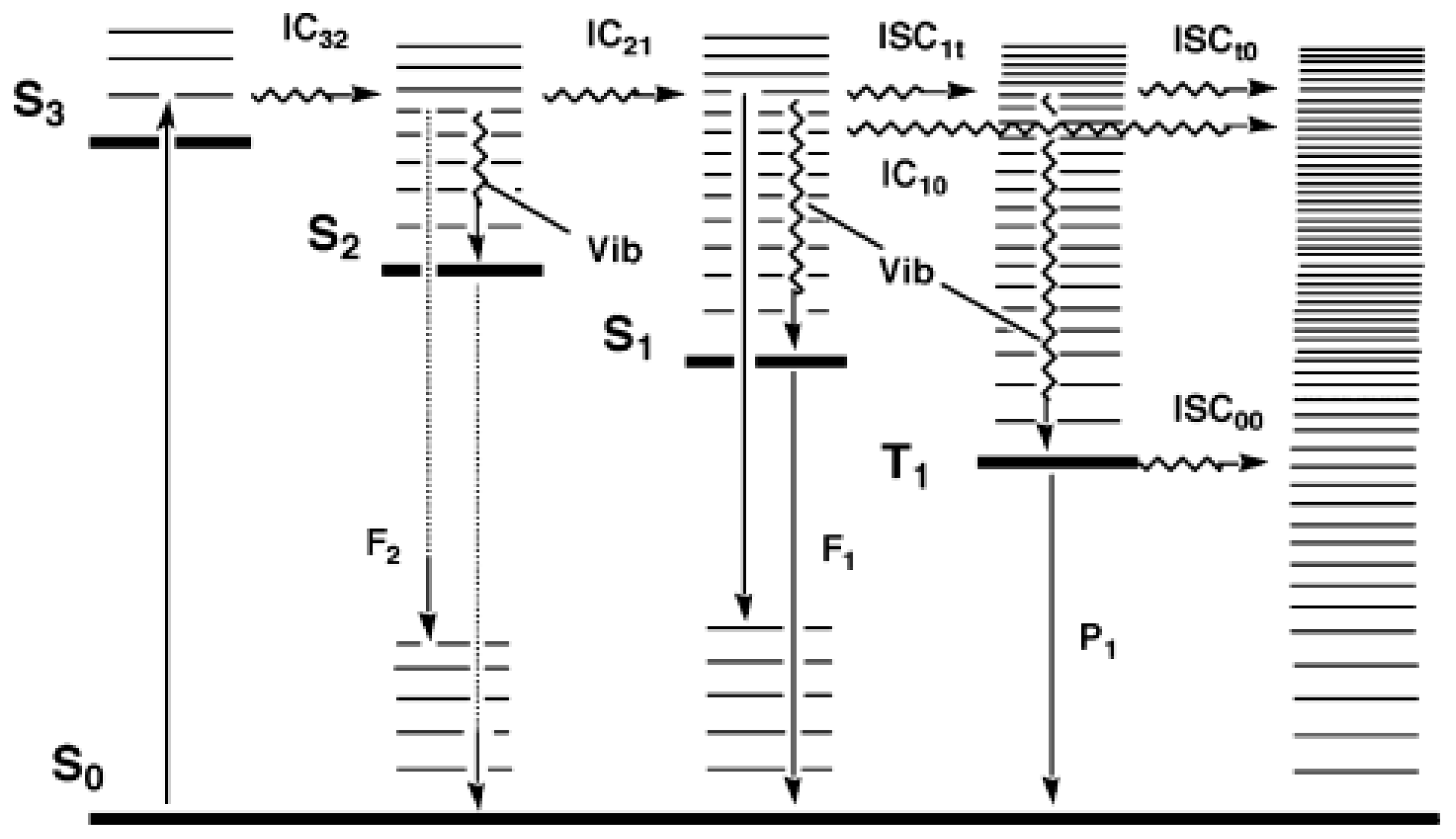
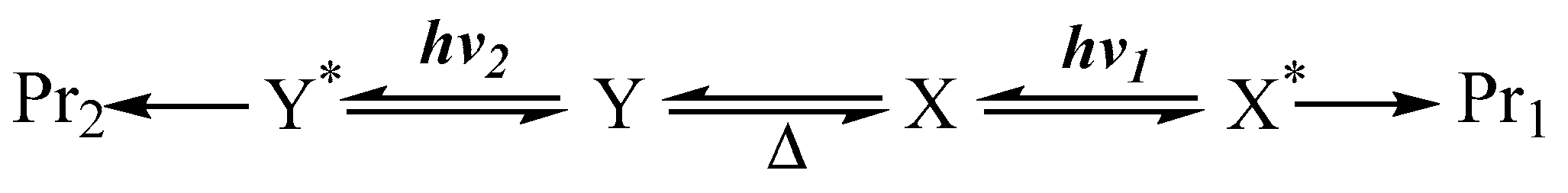
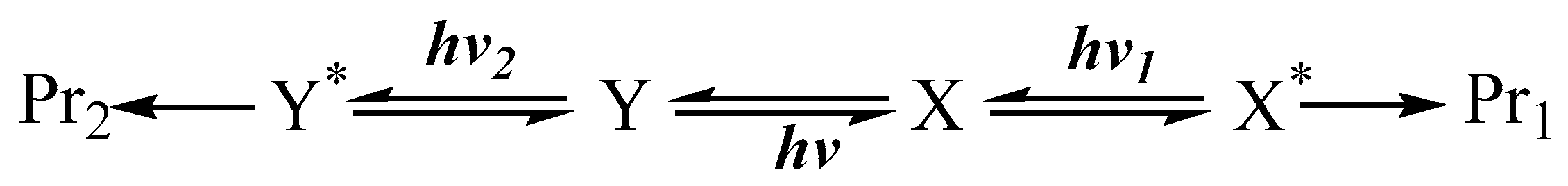
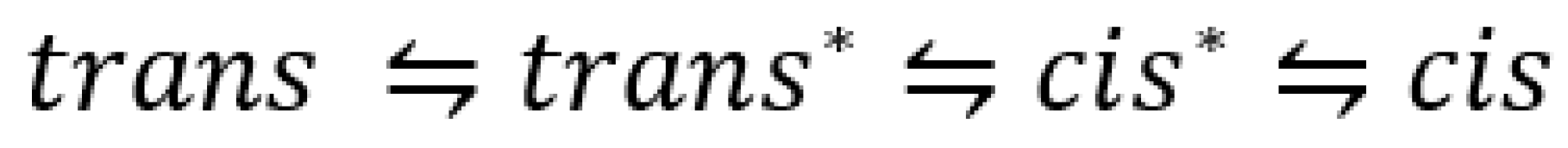
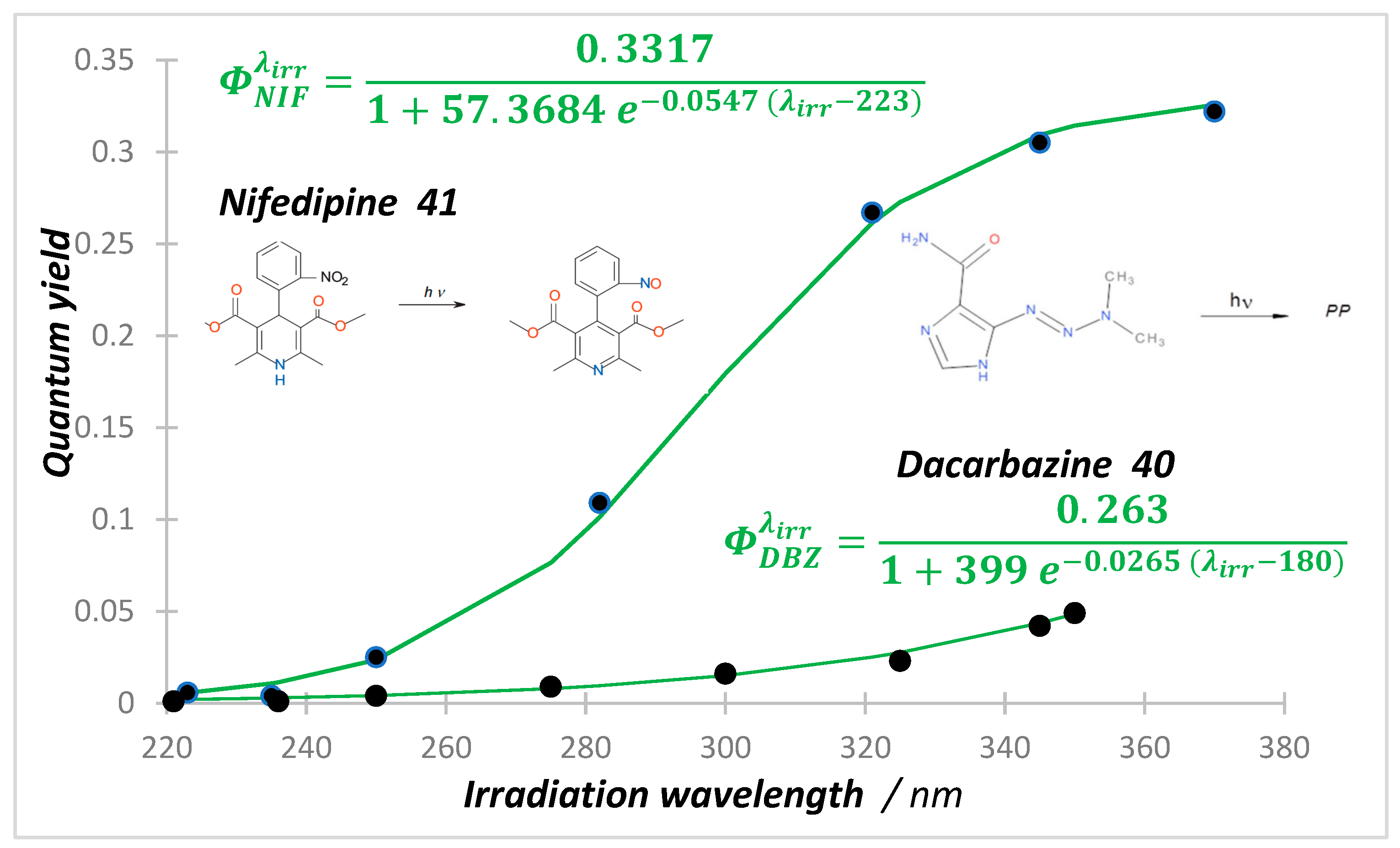
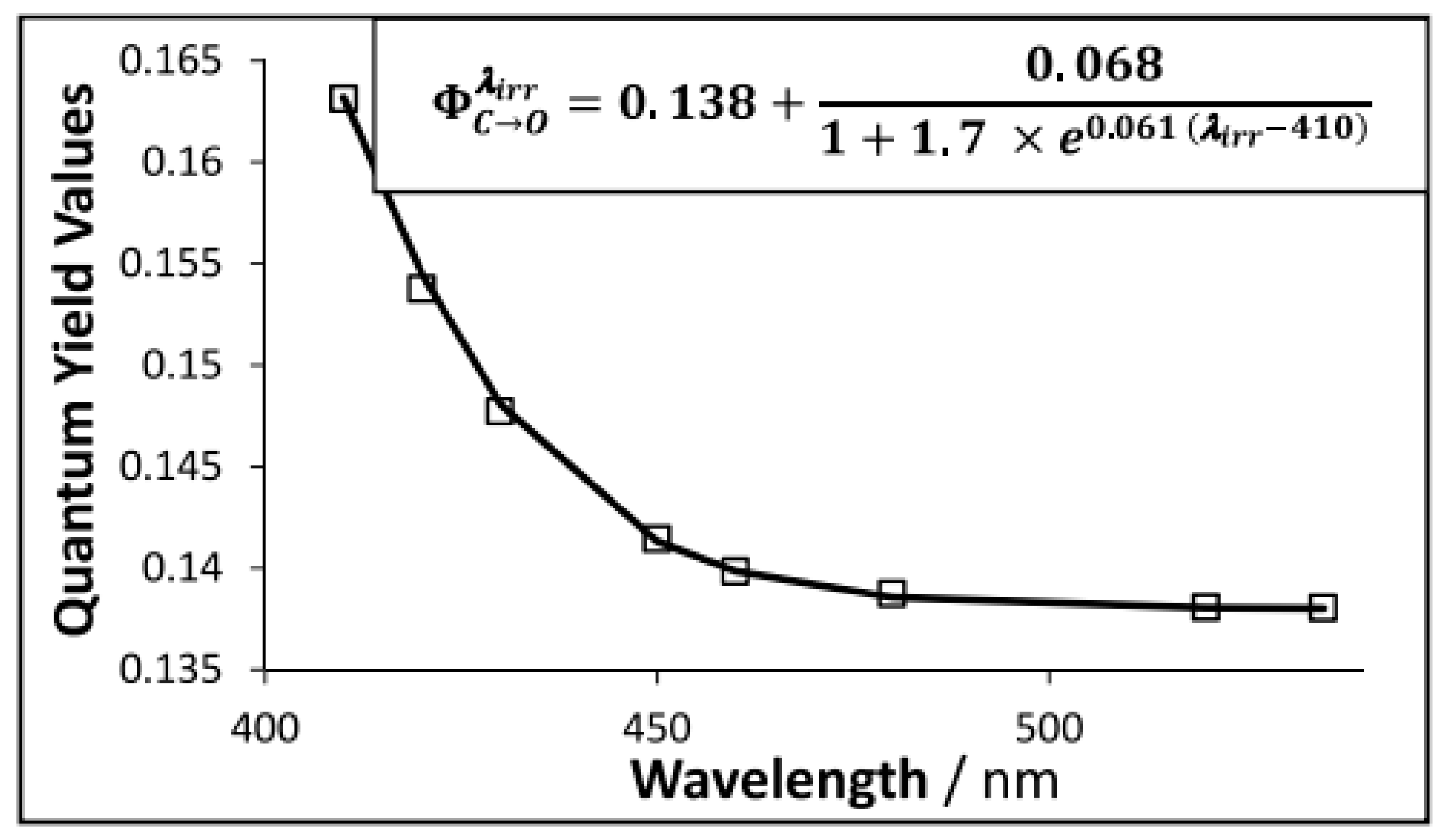
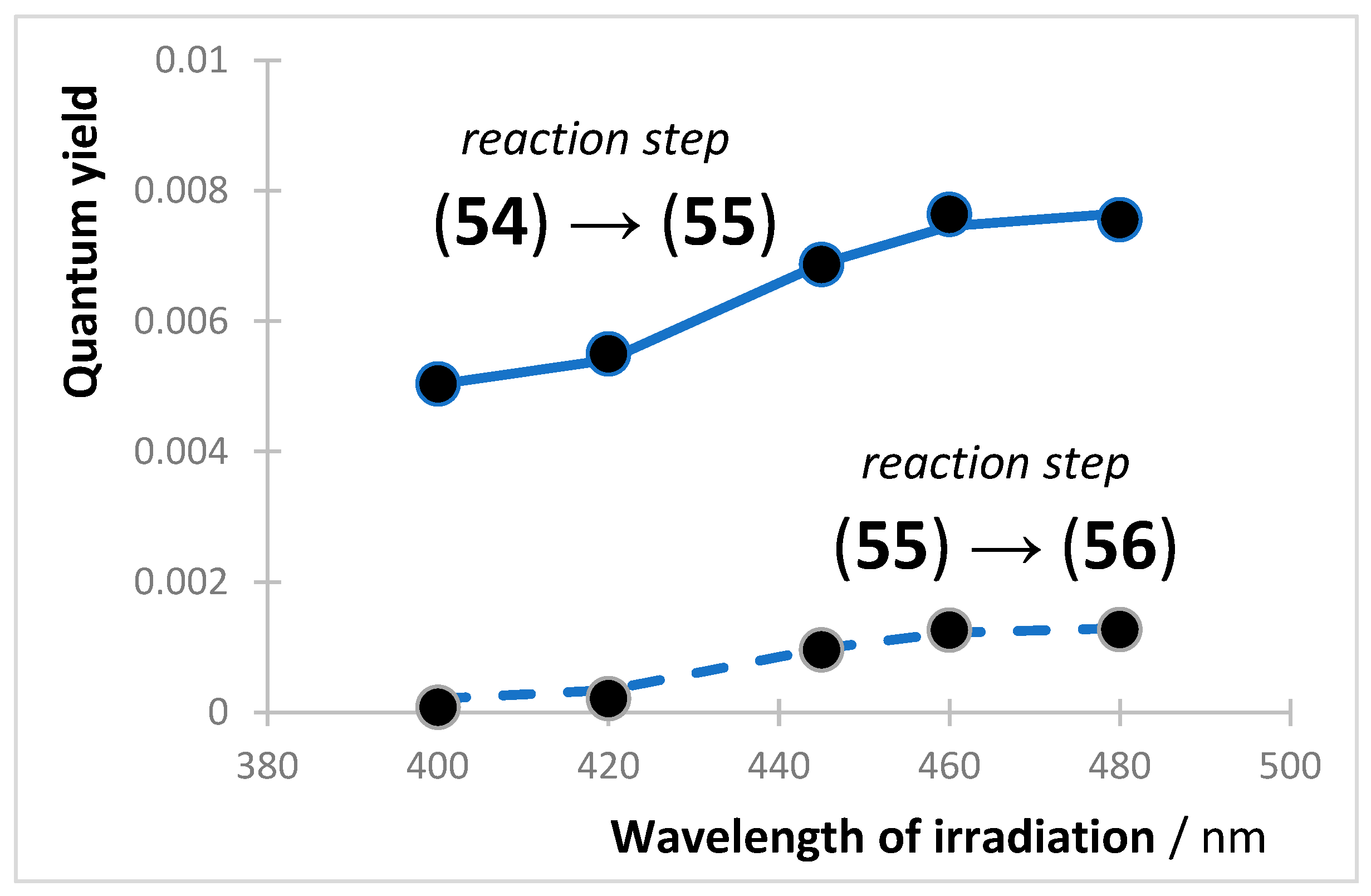
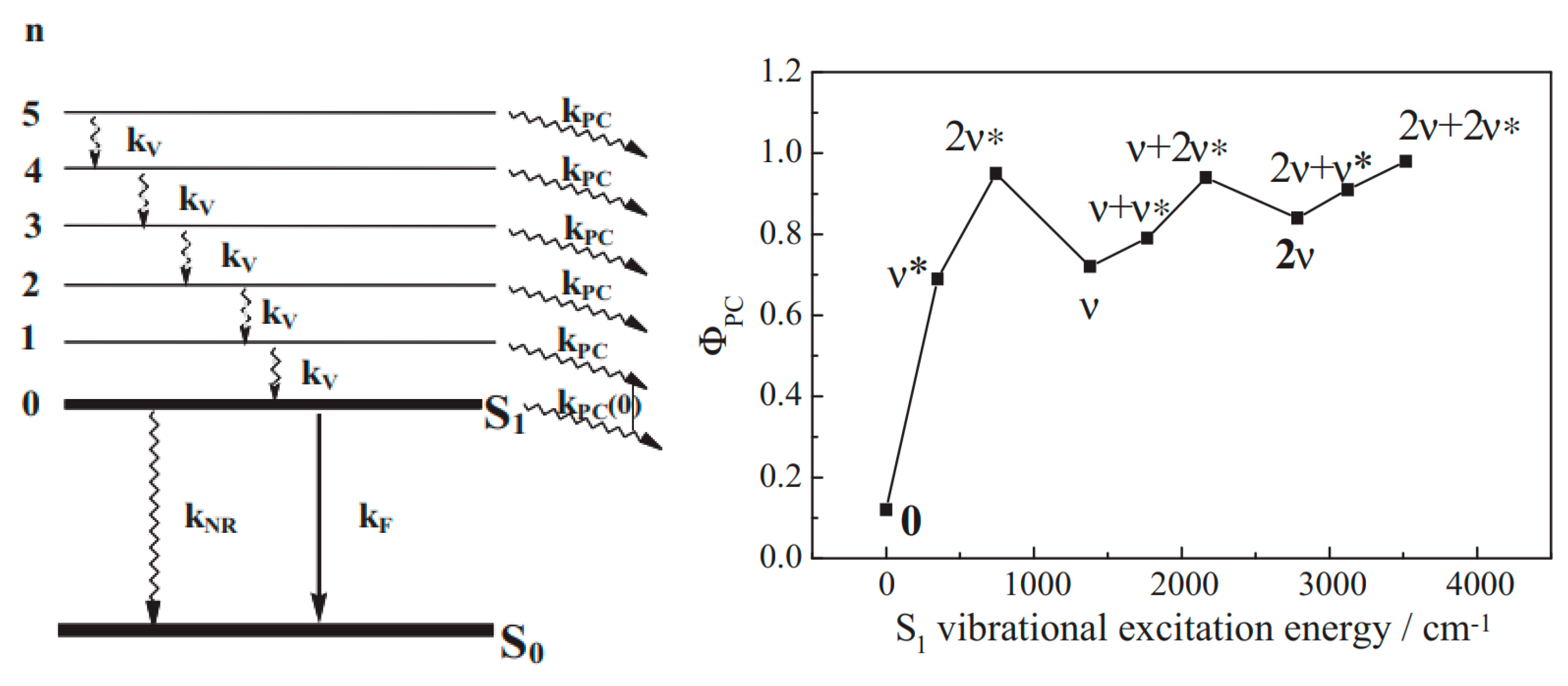
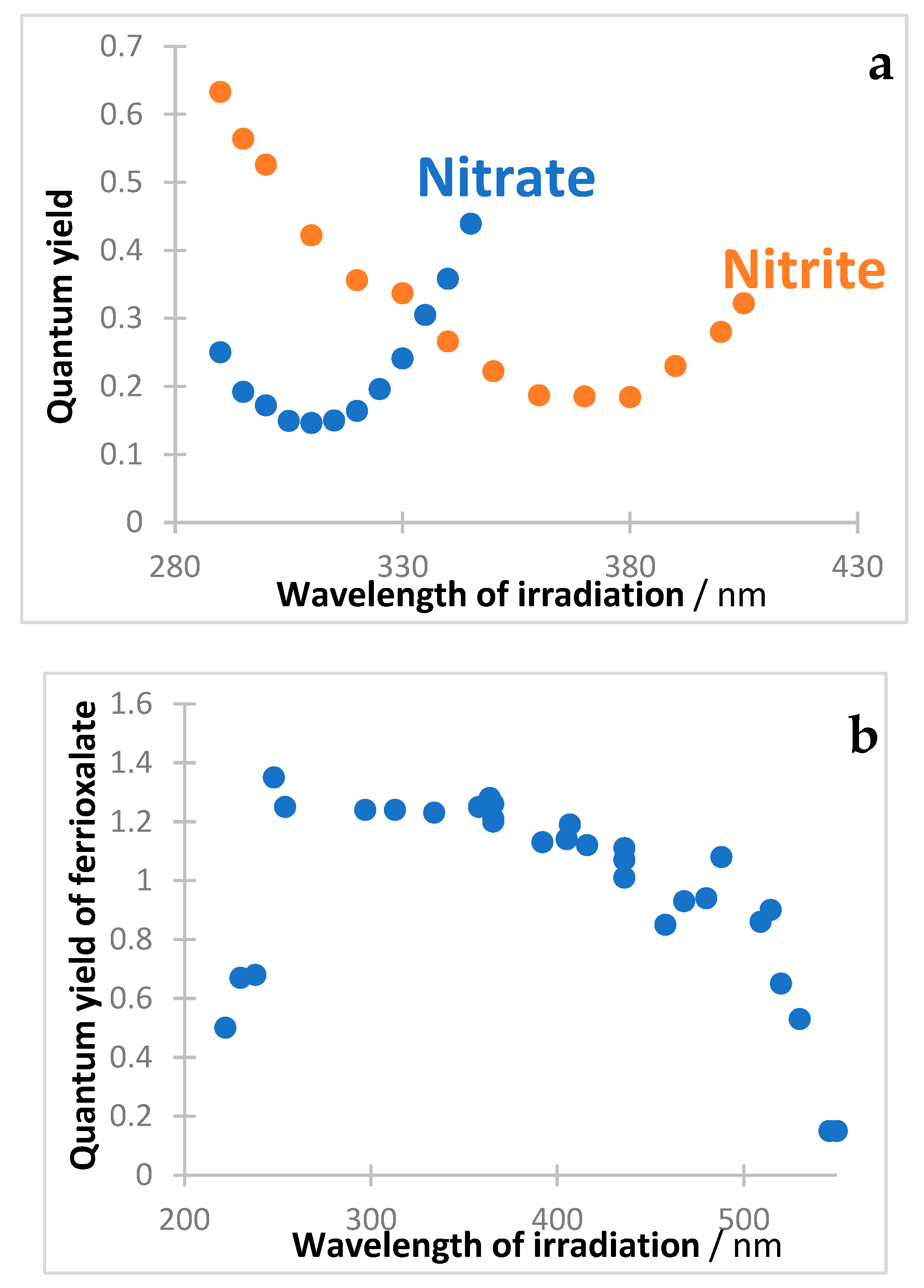
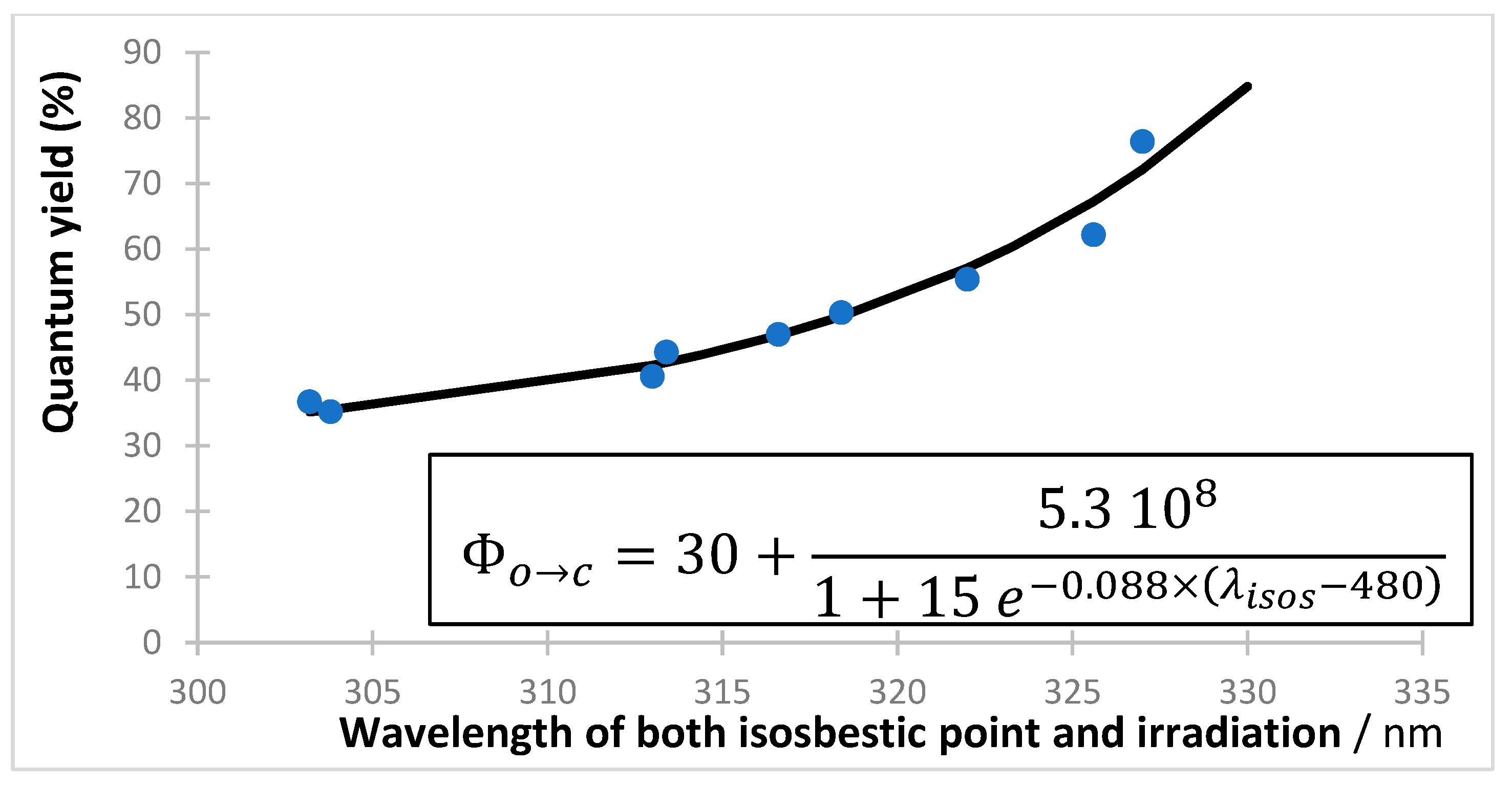
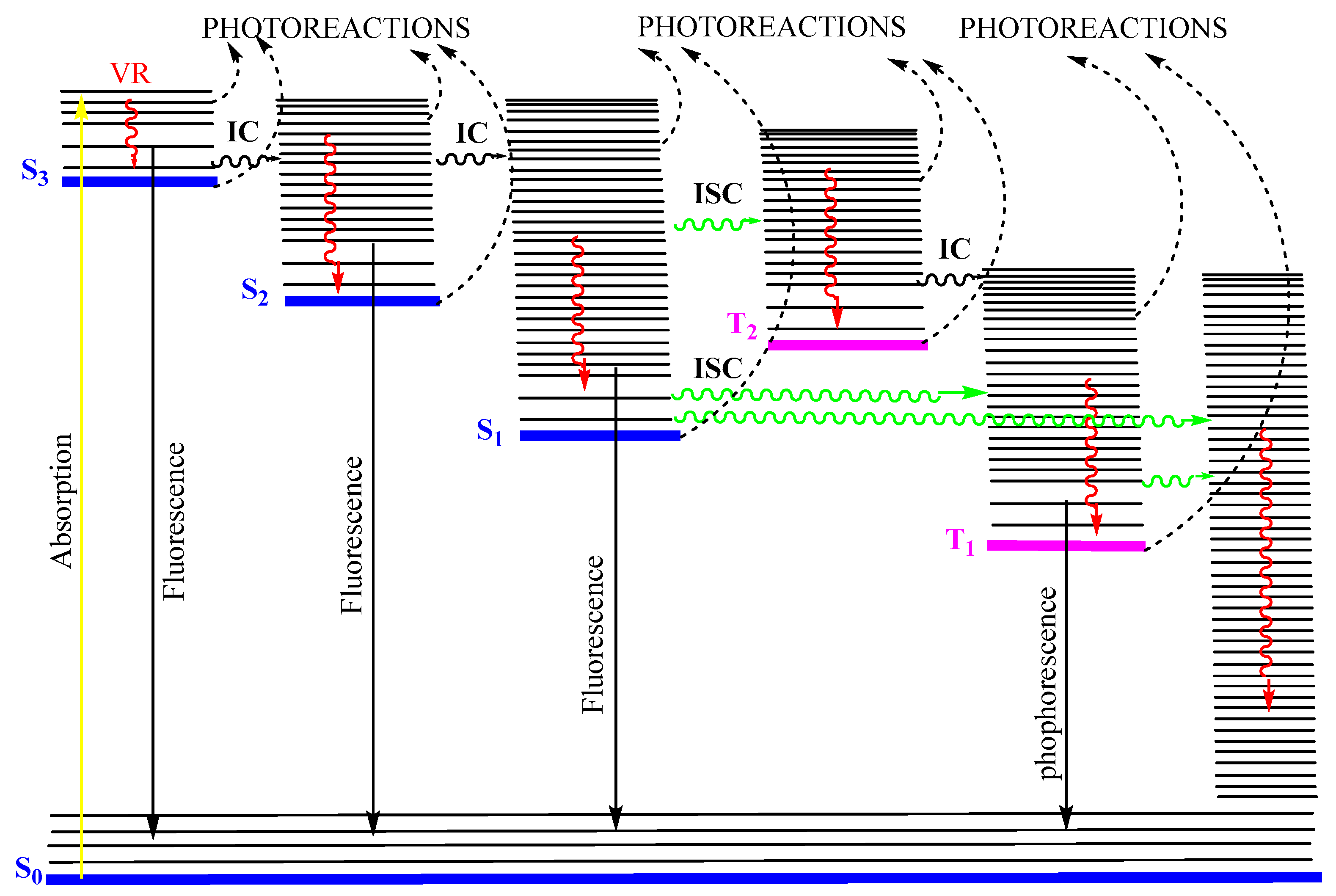
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).