Condensation By-Products in Wet Peroxide Oxidation: Fouling or Catalytic Promotion? Part I. Evidences of an Autocatalytic Process

 ,
,
Abstract
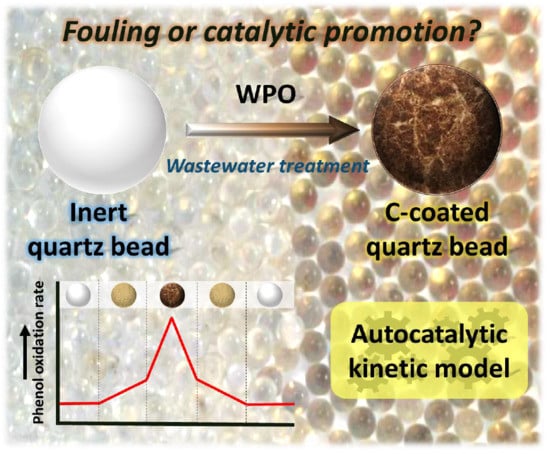
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
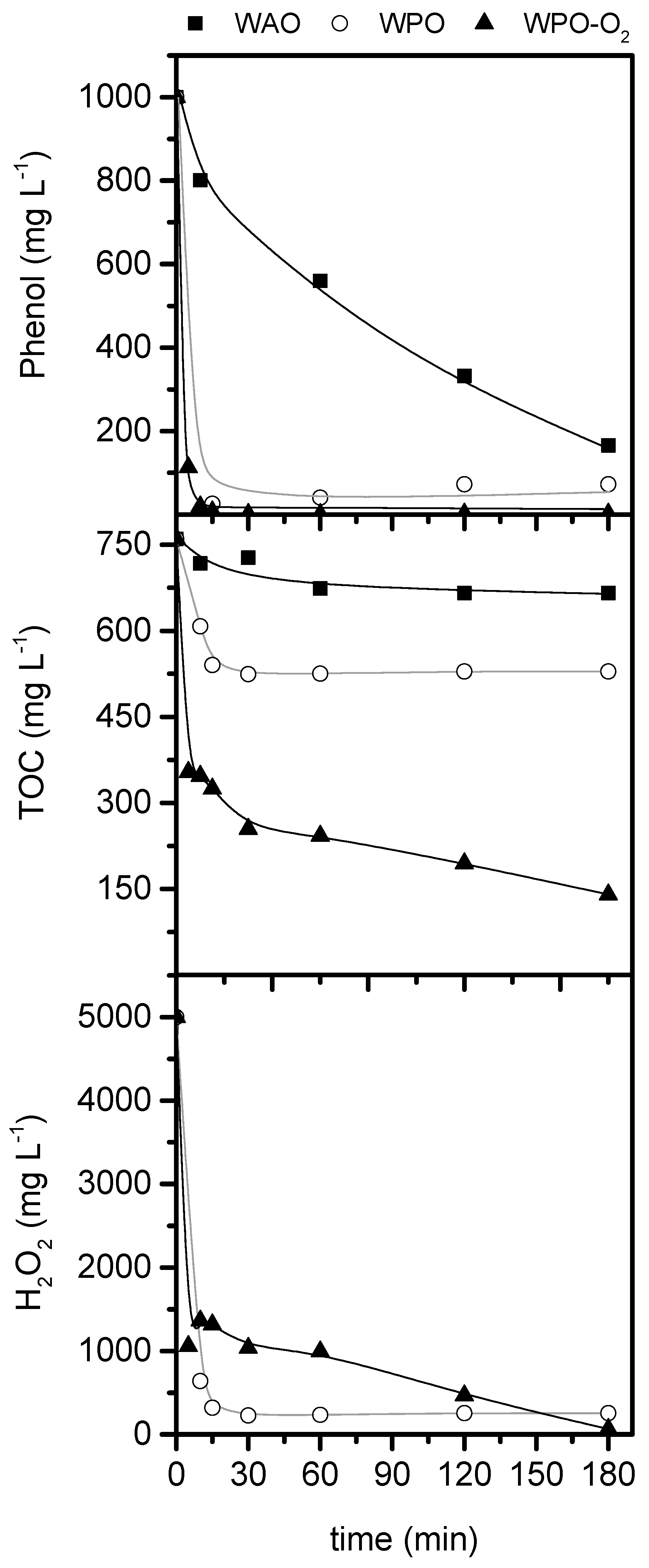
2.1. Preliminary Study
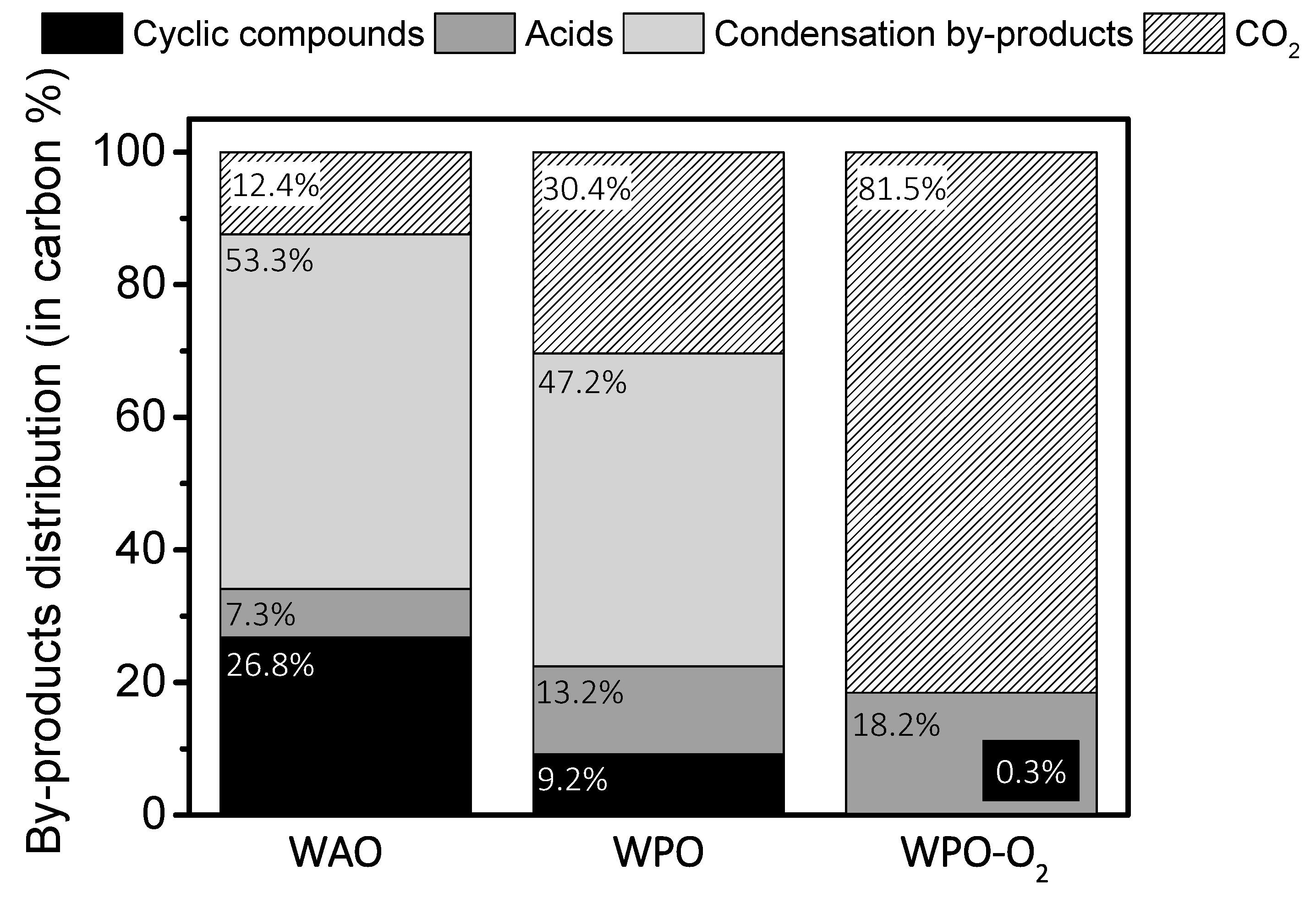
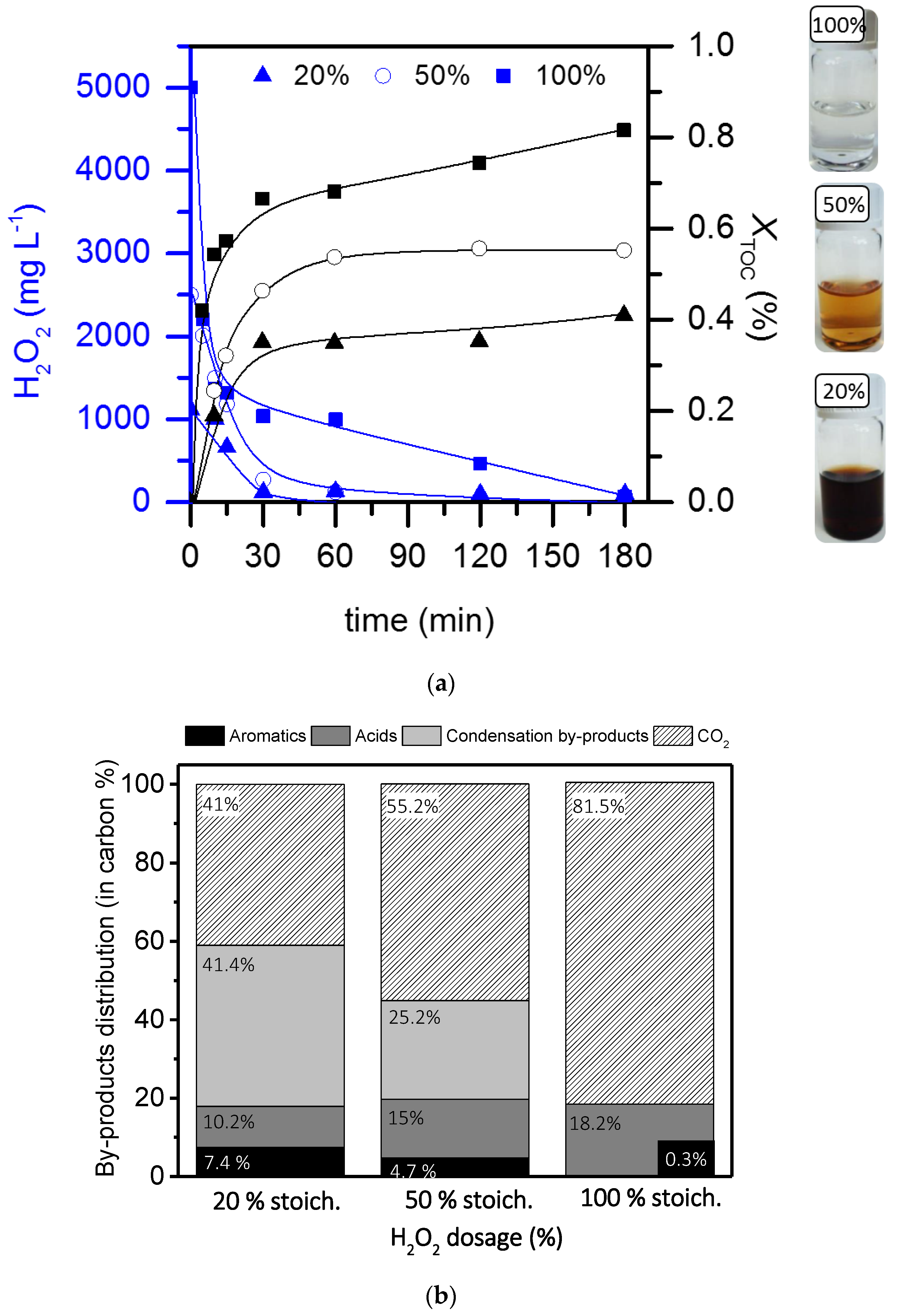
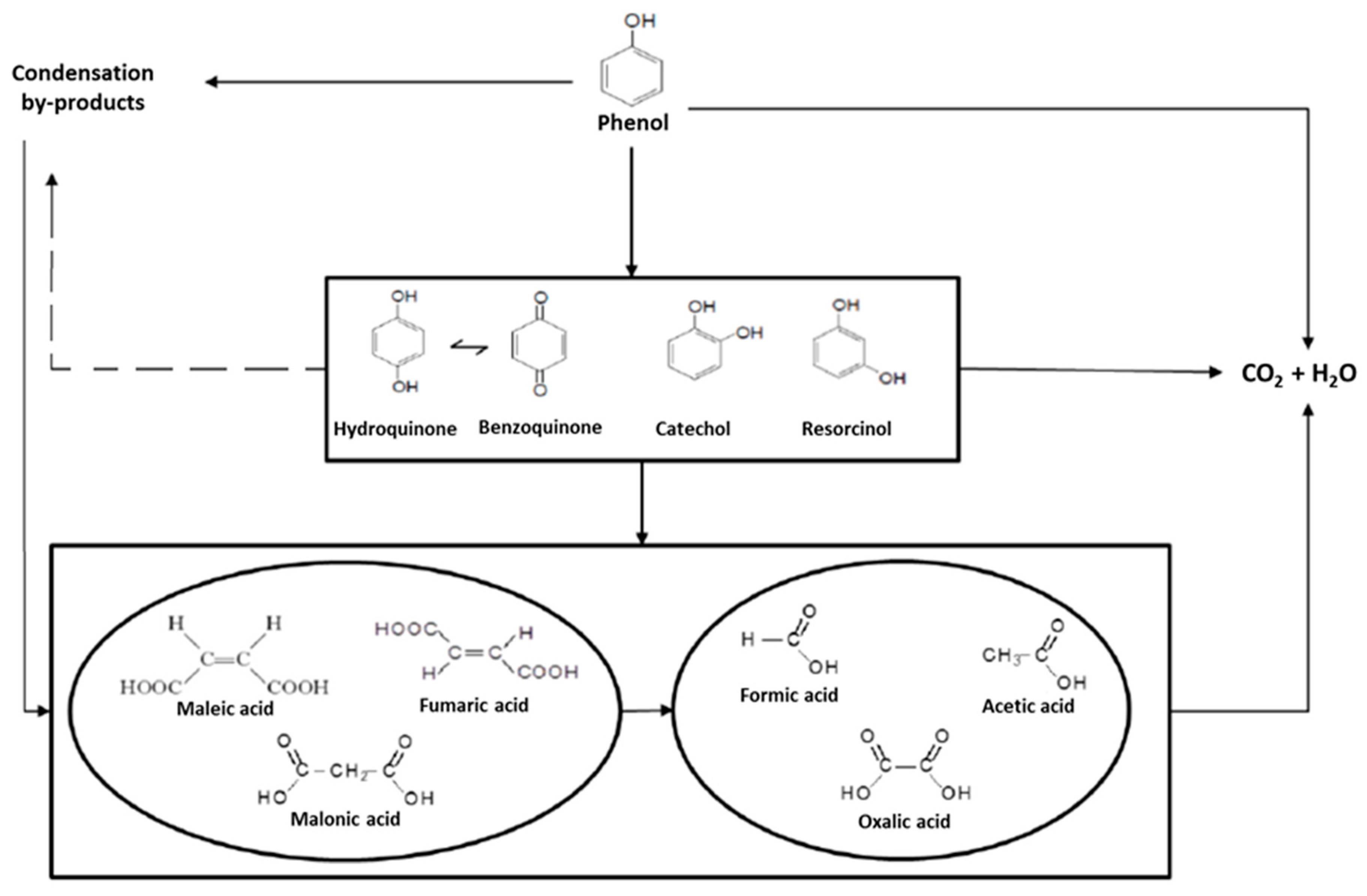
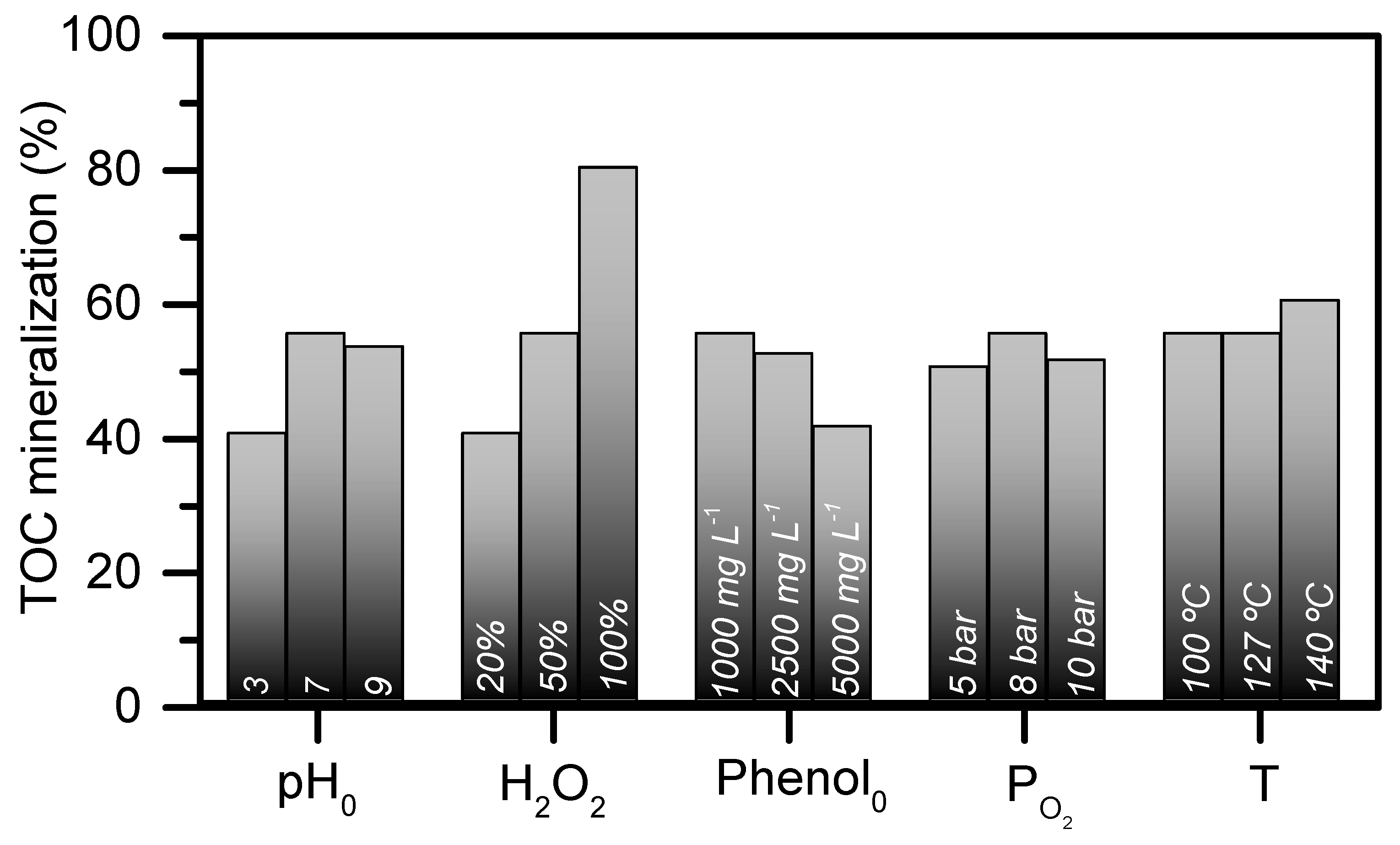
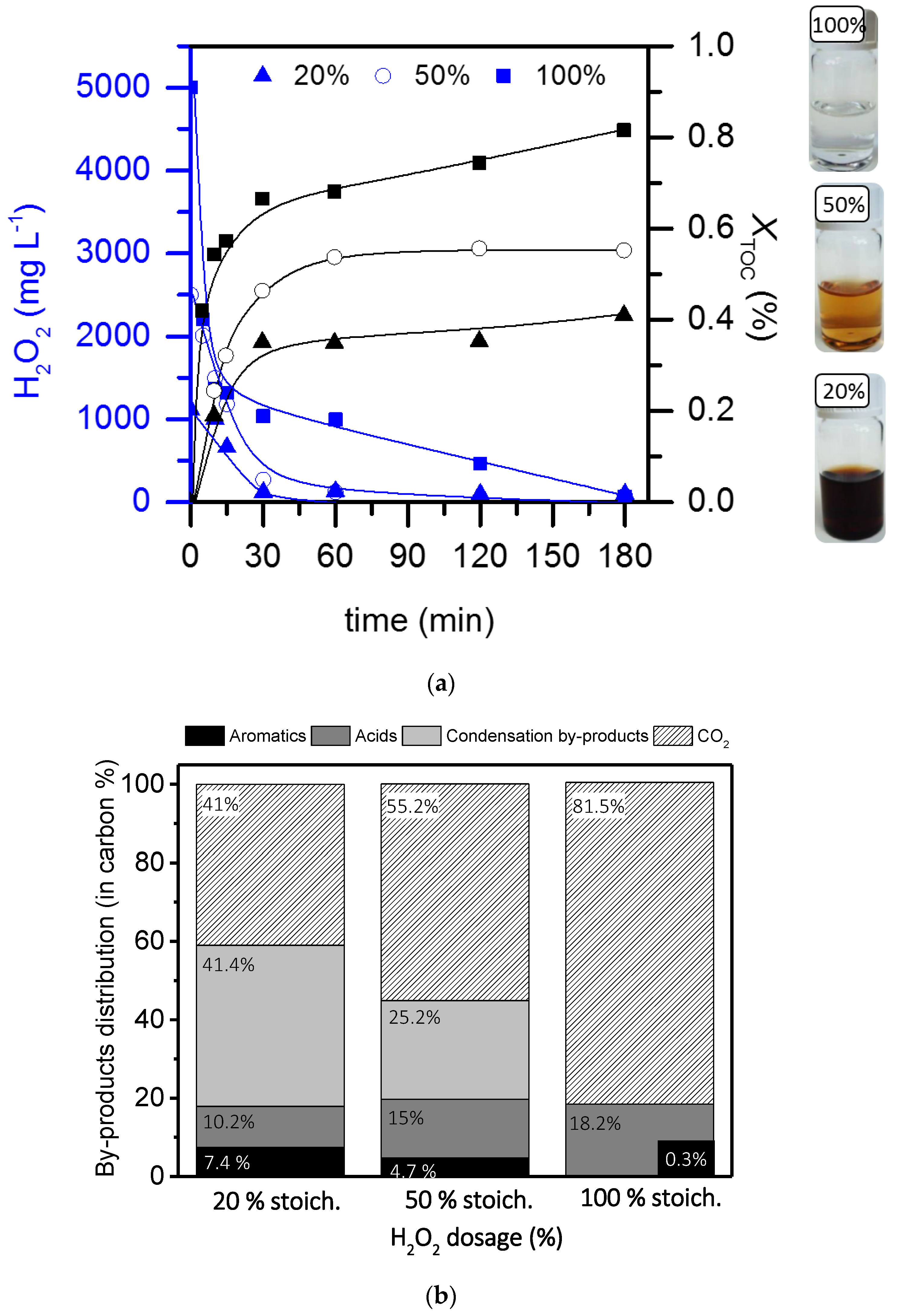
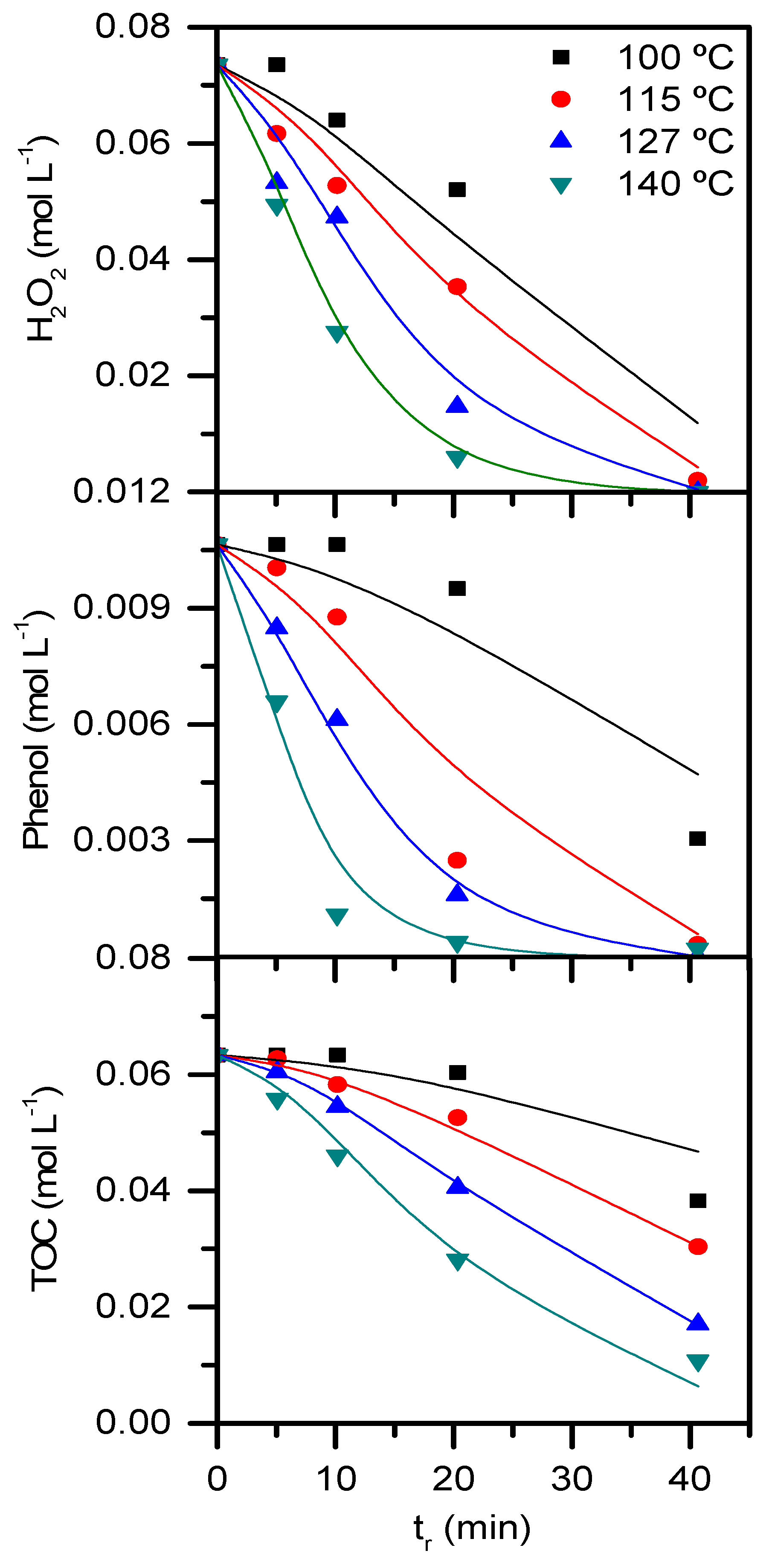
2.2. Operating conditions study: Formation of condensation by-products
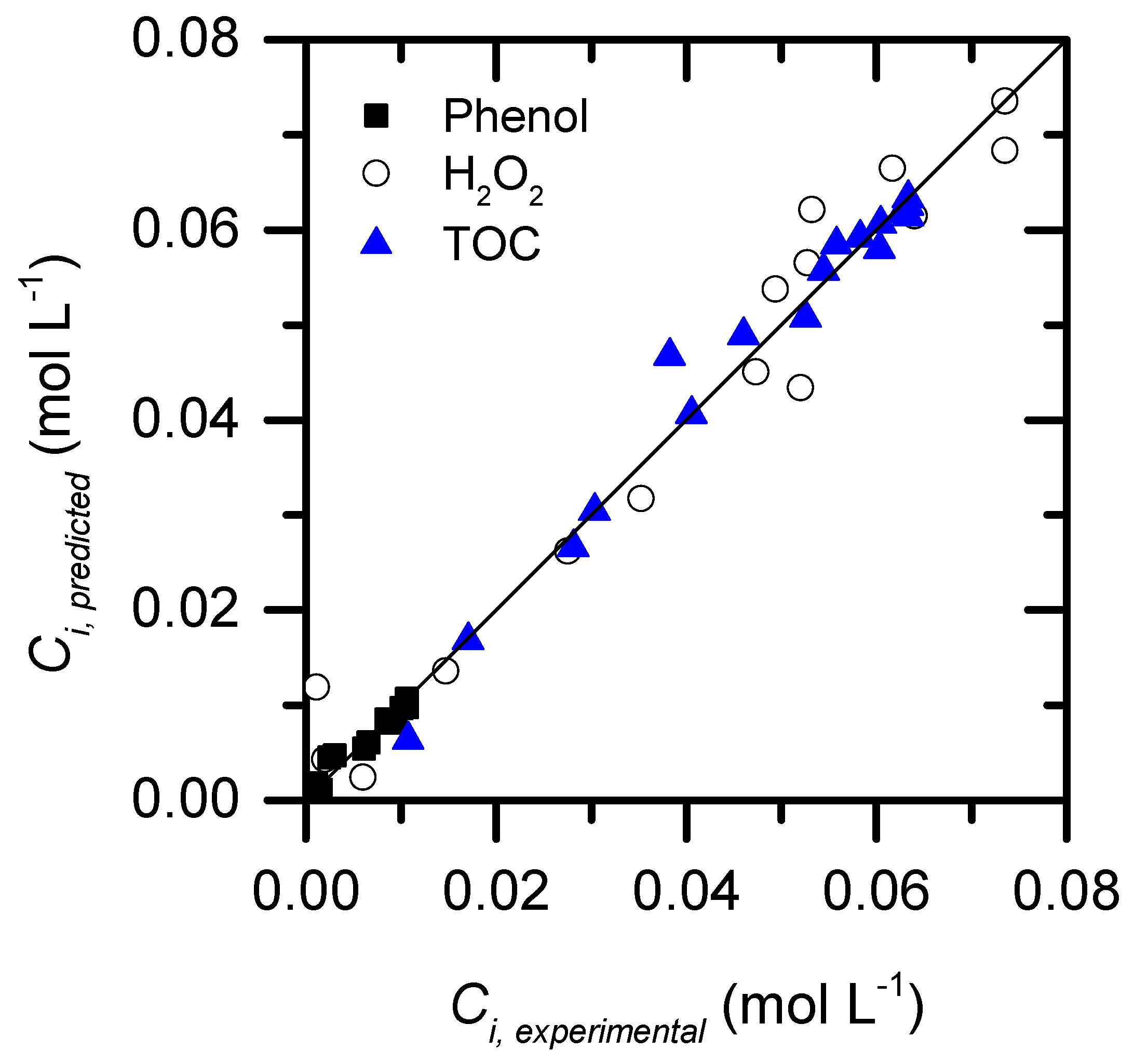
2.3. Role of condensation by-products on the kinetics of the process
3. Experimental
3.1. Oxidation experiments
3.2. Analytical methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- UN-Water 2015, Compendium of Water Quality Regulatory Frameworks: Which Water for Which Use? Available online: http://www.unwater.org/app/uploads/2017/05/Compendium-of-Water-Quality-Main-Report_4.pdf (accessed on 16 February 2019).
- Ryu, J.Y. Formation of chlorinated phenols, dibenzo-p-dioxins, dienzofurans, benzenes, benzoquinones and perchloroethylenes from phenols in oxidative and copper(II) chloride-catalyzed thermal process. Chemosphere 2008, 71, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Levec, J.; Pintar, A. Catalytic wet-air oxidation processes: A review. Catal. Today 2007, 124, 172–184. [Google Scholar] [CrossRef]
- Munoz, M.; de Pedro, Z.M.; Casas, J.A.; Rodriguez, J.J. Preparation of magnetite-based catalysts and their application in heterogeneous Fenton oxidation – A review. Appl. Catal. B 2015, 176–177, 249–265. [Google Scholar] [CrossRef]
- Zazo, J.A.; Casas, J.A.; Mohedano, A.F.; Rodríguez, J.J. Catalytic wet peroxide oxidation of phenol with a Fe/active carbon catalyst. Appl. Catal. B 2006, 65, 261–268. [Google Scholar] [CrossRef]
- Rey, A.; Faraldos, M.; Casas, J.A.; Zazo, J.A.; Bahamonde, A.; Rodríguez, J.J. Catalytic wet peroxide oxidation of phenol over Fe/AC catalysts: Influence of iron precursor and activated carbon surface. Appl. Catal. B 2009, 86, 69–77. [Google Scholar] [CrossRef]
- Keav, S.; de los Monteros, A.E.; Barbier, J.; Duprez, D. Wet Air Oxidation of phenol over Pt and Ru catalysts supported on cerium-based oxides: Resistance to fouling and kinetic modelling. Appl. Catal. B 2014, 150–151, 402–410. [Google Scholar] [CrossRef]
- Bautista, P.; Mohedano, A.F.; Menéndez, N.; Casas, J.A.; Zazo, J.A.; Rodriguez, J.J. Highly stable Fe/γAl2O3 catalyst for catalytic wet peroxide oxidation. J. Chem. Technol. Biotechnol. 2011, 86, 497–504. [Google Scholar] [CrossRef]
- Munoz, M.; Domínguez, P.; de Pedro, Z.M.; Casas, J.A.; Rodriguez, J.J. Naturally-occurring iron minerals as inexpensive catalysts for CWPO. Appl. Catal. B 2017, 203, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, C.M.; Ocón, P.; Quintanilla, A.; Casas, J.A.; Rodriguez, J.J. Highly efficient application of activated carbon as catalyst for wet peroxide oxidation. Appl. Catal. B 2013, 140–141, 663–670. [Google Scholar] [CrossRef]
- Domínguez, C.M.; Ocón, P.; Quintanilla, A.; Casas, J.A.; Rodriguez, J.J. Graphite and carbon black materials as catalysts for wet peroxide oxidation. Appl. Catal. B 2014, 144, 599–606. [Google Scholar] [CrossRef]
- Diaz de Tuesta, J.L.; Quintanilla, A.; Casas, J.A.; Rodriguez, J.J. Kinetic modeling of wet peroxide oxidation with a carbon black catalyst. Appl. Catal. B 2017, 209, 701–710. [Google Scholar] [CrossRef]
- Ribeiro, R.S.; Fathy, N.A.; Attia, A.A.; Silva, A.M.T.; Faria, J.L.; Gomes, H.T. Activated carbon xerogels for the removal of the anionic azo dyes Orange II and Chromotrope 2R by adsorption and catalytic wet peroxide oxidation. Chem. Eng. J. 2012, 195–196, 112–121. [Google Scholar] [CrossRef]
- Munoz, M.; de Pedro, Z.M.; Casas, J.A.; Rodriguez, J.J. Assessment of the generation of chlorinated byproducts upon Fenton-like oxidation of chlorophenols at different conditions. J. Hazard. Mater. 2011, 190, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Poerschmann, J.; Trommler, U.; Górecki, T.; Kopinke, F.D. Formation of chlorinated biphenyls, diphenyl ethers and benzofurans as a result of Fenton-driven oxidation of 2-chlorophenol. Chemosphere 2009, 75, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Poerschmann, J.; Trommler, U.; Górecki, T. Aromatic intermediate formation during oxidative degradation of Bisphenol A by homogeneous sub-stoichiometric Fenton reaction. Chemosphere 2010, 79, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, M.; Fernández-Castro, P.; San Román, M.F.; Ortiz, I. Assessment of PCDD/Fs formation in the Fenton oxidation of 2-chlorophenol: Influence of the iron dose applied. Chemosphere 2015, 137, 135–141. [Google Scholar] [CrossRef]
- Vallejo, M.; Fresnedo San Román, M.; Ortiz, I.; Irabien, A. Overview of the PCDD/Fs degradation potential and formation risk in the application of advanced oxidation processes (AOPs) to wastewater treatment. Chemosphere 2015, 118, 44–56. [Google Scholar] [CrossRef]
- Delgado, J.J.; Pérez-Omil, J.A.; Rodríguez-Izquierdo, J.M.; Cauqui, M.A. The role of the carbonaceous deposits in the Catalytic Wet Oxidation (CWO) of phenol. Catal. Comm. 2006, 7, 639–643. [Google Scholar] [CrossRef]
- Delgado, J.J.; Chen, X.; Pérez-Omil, J.A.; Rodríguez-Izquierdo, J.M.; Cauqui, M.A. The effect of reaction conditions on the apparent deactivation of Ce–Zr mixed oxides for the catalytic wet oxidation of phenol. Catal. Today 2012, 180, 25–33. [Google Scholar] [CrossRef]
- Lee, D.K.; Kim, D.S.; Kim, T.H.; Lee, Y.K.; Jeong, S.E.; Le, N.T.; Cho, M.J.; Henam, S.D. Deactivation of Pt catalysts during wet oxidation of phenol. Catal. Today 2010, 154, 244–249. [Google Scholar] [CrossRef]
- Collett, C.H.; McGregor, J. Things go better with coke: the beneficial role of carbonaceous deposits in heterogeneous catalysis. Catal. Sci. Technol. 2016, 6, 363–378. [Google Scholar] [CrossRef]
- Gornay, J.; Coniglio, L.; Billaud, F.; Wild, G. Octanoic acid pyrolysis in a stainless-steel tube: What is the role of the coke formed on the wall? J. Anal. Appl. Pyrol. 2010, 87, 78–84. [Google Scholar] [CrossRef]
- Teschner, D.; Vass, E.; Hävecker, M.; Zafeiratos, S.; Schnörch, P.; Sauer, H.; Knop-Gericke, A.; Schlögl, R.; Chamam, M.; Wootsch, A.; et al. Alkyne hydrogenation over Pd catalysts: A new paradigm. J. Catal. 2006, 242, 26–37. [Google Scholar] [CrossRef]
- Vrieland, G.E.; Menon, P.G. Nature of the catalytically active carbonaceous sites for the oxydehydrogenation of ethylbenzene to styrene: A brief review. Appl. Catal. 1991, 77, 1–8. [Google Scholar] [CrossRef]
- Fiedorow, R.; Przystajko, W.; Sopa, M.; Dalla Lana, I.G. The nature and catalytic influence of coke formed on alumina: Oxidative dehydrogenation of ethylbenzene. J. Catal. 1981, 68, 33–41. [Google Scholar] [CrossRef]
- Hamoudi, S.; Larachi, F.; Adnot, A.; Sayari, A. Characterization of Spent MnO2/CeO2 Wet Oxidation Catalyst by TPO–MS, XPS, and S-SIMS. J. Catal. 1999, 185, 333–344. [Google Scholar] [CrossRef]
- Kim, S.; Ihm, S. Nature of carbonaceous deposits on the alumina supported transition metal oxide catalysts in the wet air oxidation of phenol. Top. Catal. 2005, 33, 171–179. [Google Scholar] [CrossRef]
- Catrinescu, C.; Teodosiu, C.; Macoveanu, M.; Miehe-Brendlé, J.; Le Dred, R. Catalytic wet peroxide oxidation of phenol over Fe-exchanged pillared beidellite. Water Res. 2003, 37, 1154–1160. [Google Scholar] [CrossRef]
- Yadav, A.; Verma, N. Carbon bead-supported copper-dispersed carbon nanofibers: An efficient catalyst for wet air oxidation of industrial wastewater in a recycle flow reactor. J. Ind. Eng. Chem. 2018, 67, 448–460. [Google Scholar] [CrossRef]
- Huang, H.; Lu, M.; Chen, J. Catalytic Decomposition of hydrogen peroxide and 2-chlorophenol with iron oxides. Water Res. 2001, 35, 2291–2299. [Google Scholar] [CrossRef]
- Pham, A.L.; Lee, C.; Doyle, F.M.; Sedlak, D.L. A silica-supported iron oxide catalyst capable of activating hydrogen peroxide at neutral pH values. Environ. Sci. Technol. 2009, 43, 8930–8935. [Google Scholar] [CrossRef]
- Kwon, B.G.; Lee, D.S.; Kang, N.; Yoon, J. Characteristics of p-chlorophenol oxidation by Fenton’s reagent. Water Res. 1999, 33, 2110–2118. [Google Scholar] [CrossRef]
- Munoz, M.; de Pedro, Z.M.; Casas, J.A.; Rodriguez, J.J. Combining efficiently catalytic hydrodechlorination and wet peroxide oxidation (HDC–CWPO) for the abatement of organochlorinated water pollutants. Appl. Catal. B 2014, 150–151, 197–203. [Google Scholar] [CrossRef]
- Mijangos, F.; Varona, F.; Villota, N. Changes in solution color during phenol oxidation by Fenton reagent. Environ. Sci. Technol. 2006, 40, 5538–5543. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.D.; Hamilton, G.A. CHAPTER II - Mechanisms of phenolic oxidative coupling reactions. Org. Chem. 1973, 5, 97–134. [Google Scholar]
- Garcia-Costa, A.L.; Zazo, J.A.; Rodriguez, J.J.; Casas, J.A. Microwave-assisted catalytic wet peroxide oxidation. Comparison of Fe catalysts supported on activated carbon and -alumina. Appl. Catal. B 2017, 218, 637–642. [Google Scholar] [CrossRef]
- Quintanilla, A.; Casas, J.A.; Zazo, J.A.; Mohedano, A.F.; Rodríguez, J.J. Wet air oxidation of phenol at mild conditions with a Fe/activated carbon catalyst. Appl. Catal. B 2006, 62, 115–120. [Google Scholar] [CrossRef]
- Eisenberg, G.M. Colorimetric determination of hydrogen peroxide. Ind. Eng. Chem. Res. 1943, 15, 327–328. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintanilla, A.; Diaz de Tuesta, J.L.; Figueruelo, C.; Munoz, M.; Casas, J.A. Condensation By-Products in Wet Peroxide Oxidation: Fouling or Catalytic Promotion? Part I. Evidences of an Autocatalytic Process. Catalysts 2019, 9, 516. https://doi.org/10.3390/catal9060516
Quintanilla A, Diaz de Tuesta JL, Figueruelo C, Munoz M, Casas JA. Condensation By-Products in Wet Peroxide Oxidation: Fouling or Catalytic Promotion? Part I. Evidences of an Autocatalytic Process. Catalysts. 2019; 9(6):516. https://doi.org/10.3390/catal9060516
Chicago/Turabian StyleQuintanilla, Asunción, Jose L. Diaz de Tuesta, Cristina Figueruelo, Macarena Munoz, and Jose A. Casas. 2019. "Condensation By-Products in Wet Peroxide Oxidation: Fouling or Catalytic Promotion? Part I. Evidences of an Autocatalytic Process" Catalysts 9, no. 6: 516. https://doi.org/10.3390/catal9060516