



Animals 2024, 14(20), 3026; https://doi.org/10.3390/ani14203026 - 19 Oct 2024
Viewed by 708
Abstract
►
Show Figures
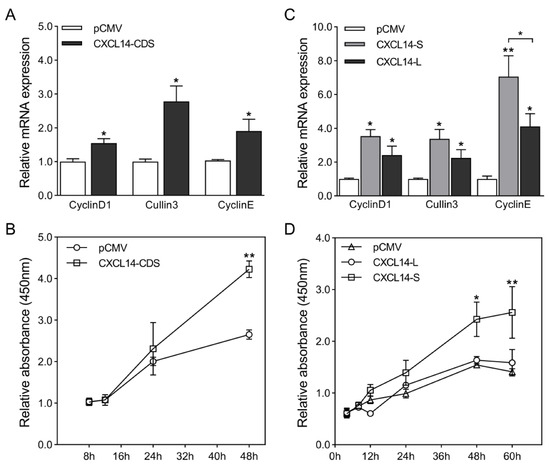
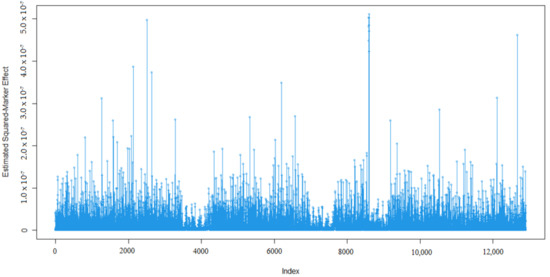
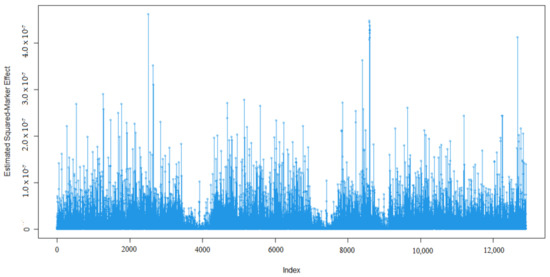
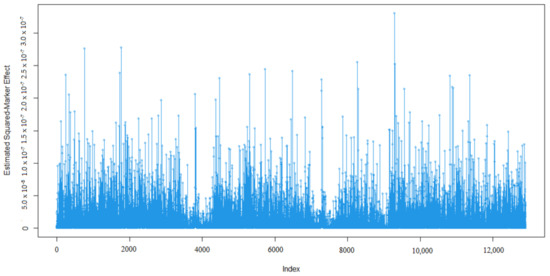
Heat stress severely affects dairy cattle production and reproduction performances in tropical regions. Genetic selection to maintain adequate yield and reproductive performance while enhancing their ability to withstand heat is essential for improving the genetics of dairy cows. Therefore, in this study, we
[...] Read more.
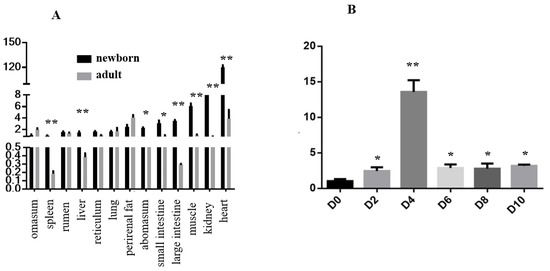
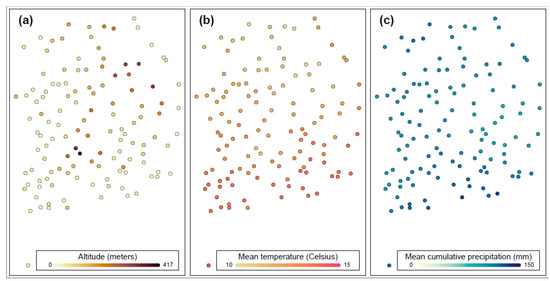
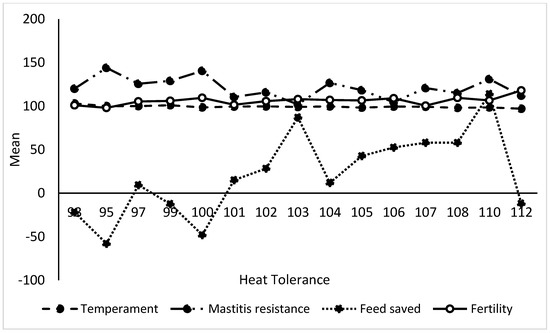
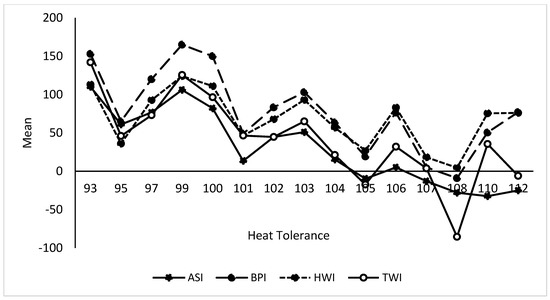
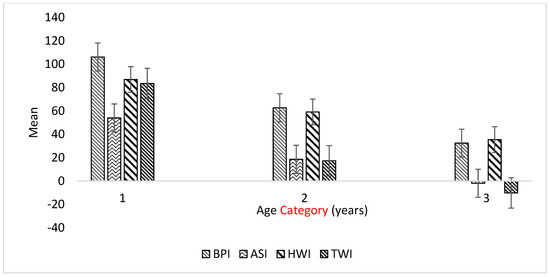
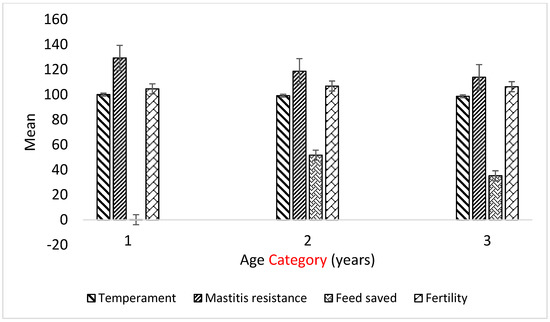
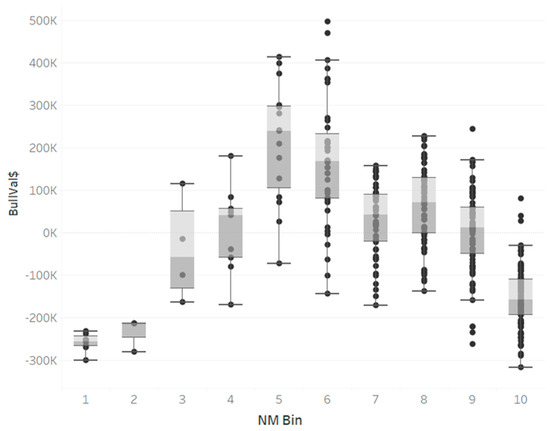
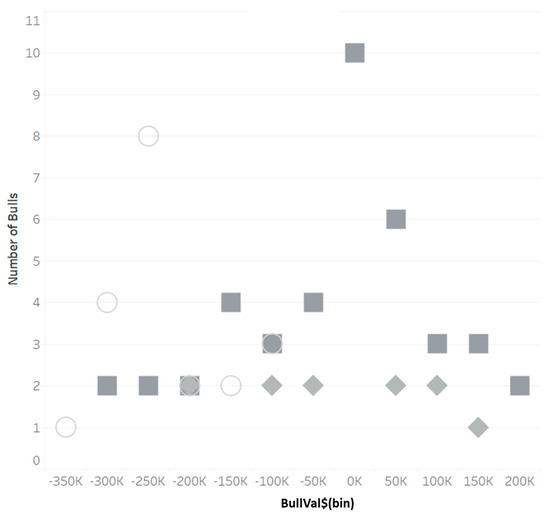
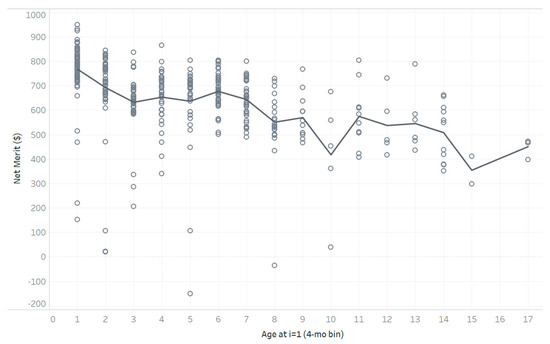
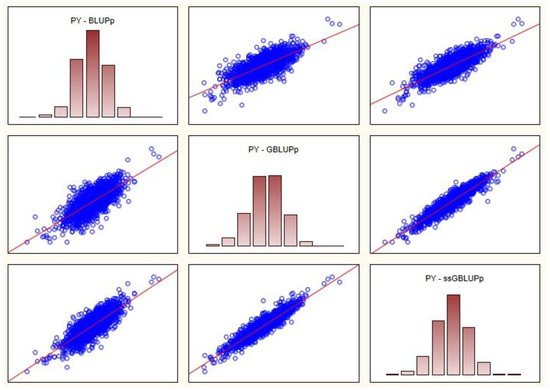
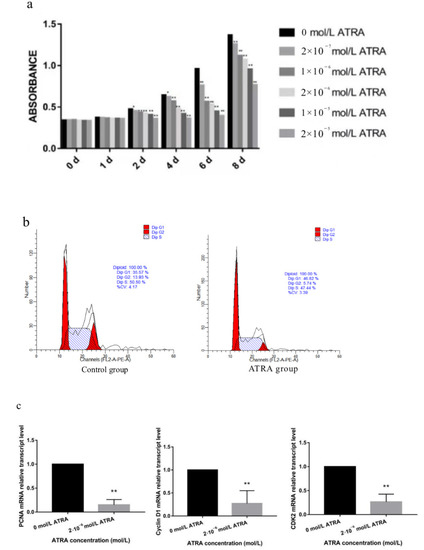
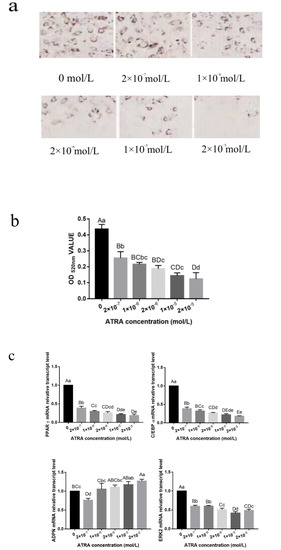
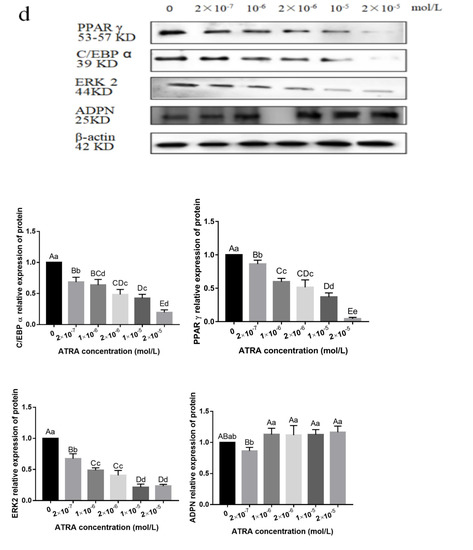
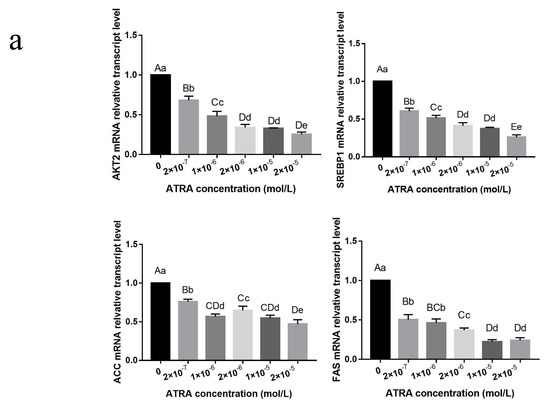
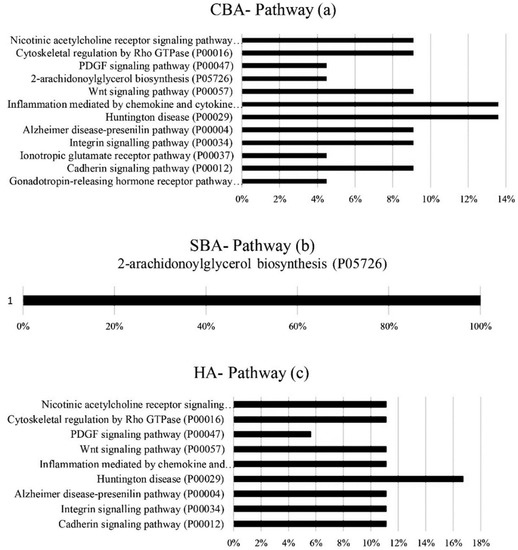
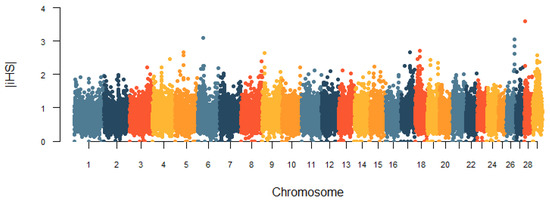
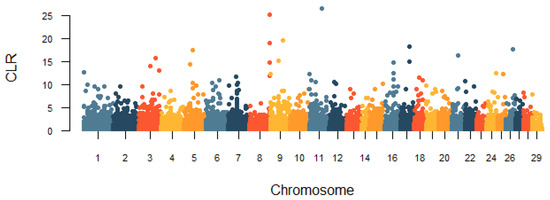
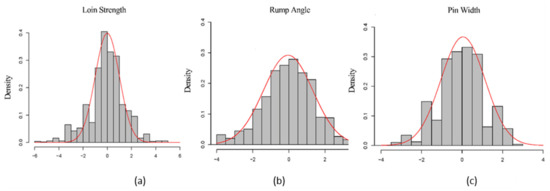
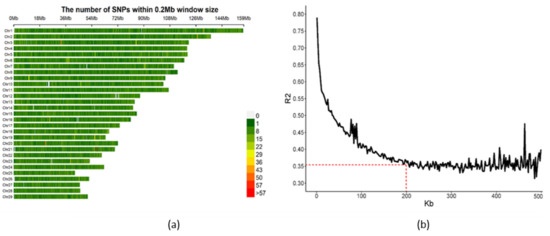
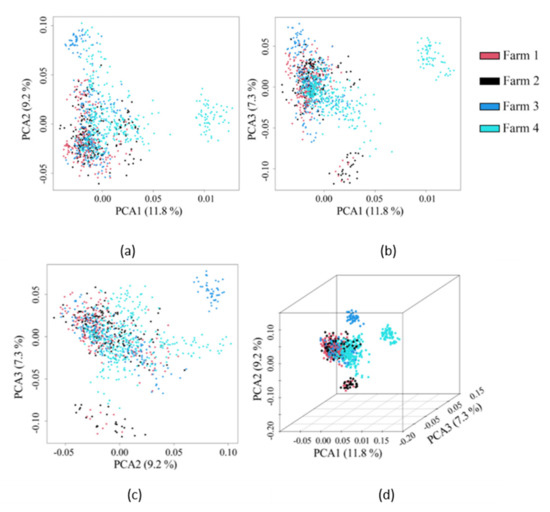
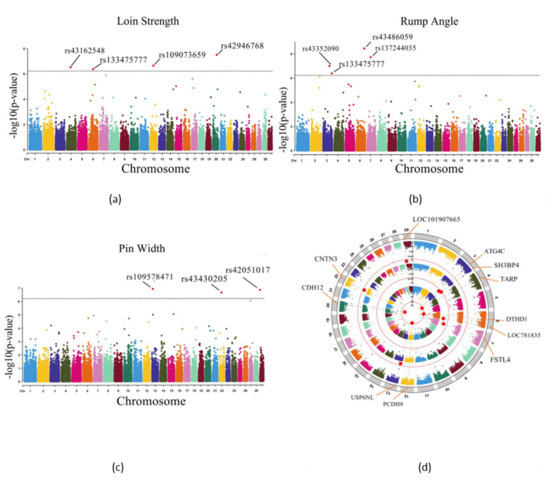
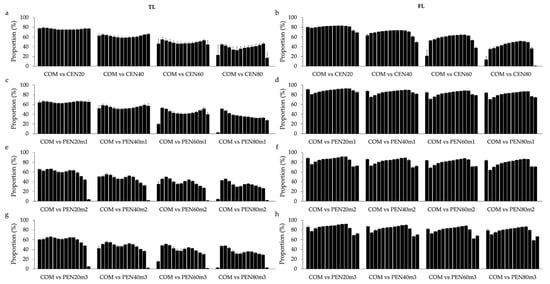
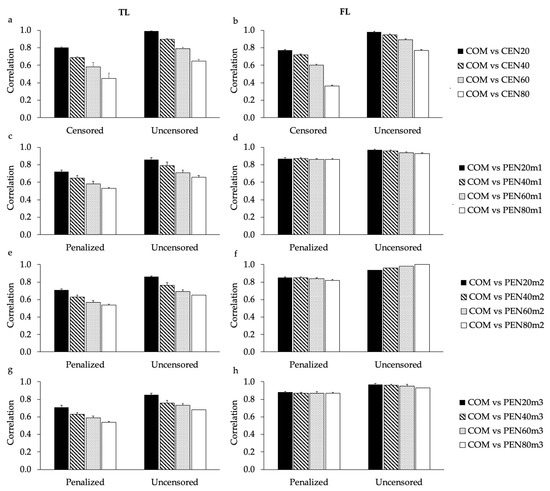
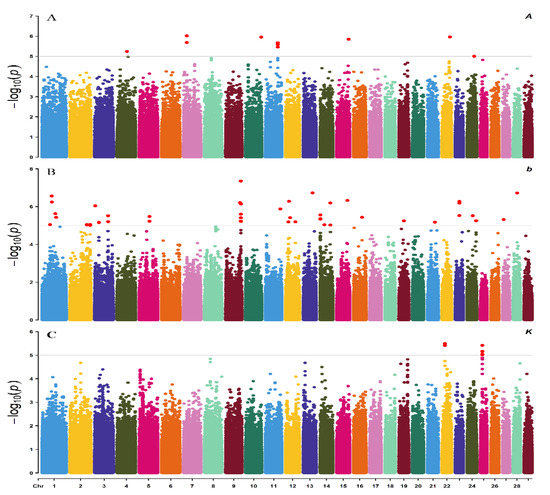
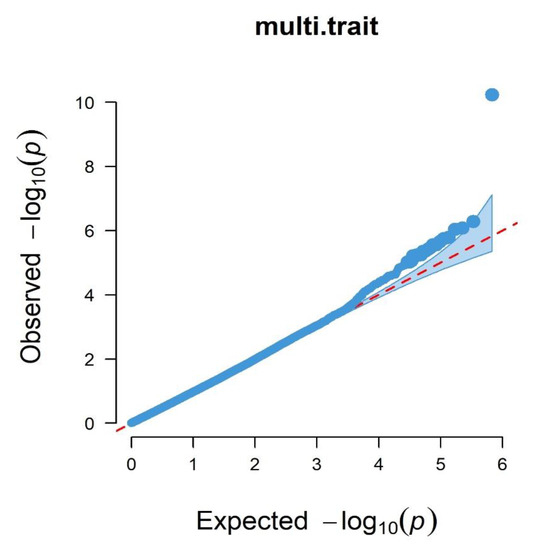
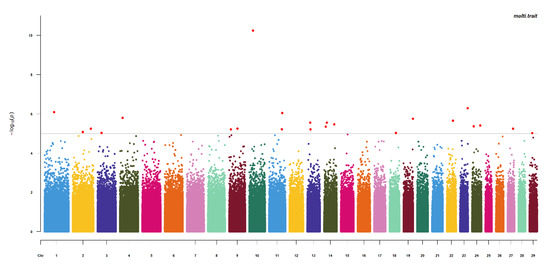
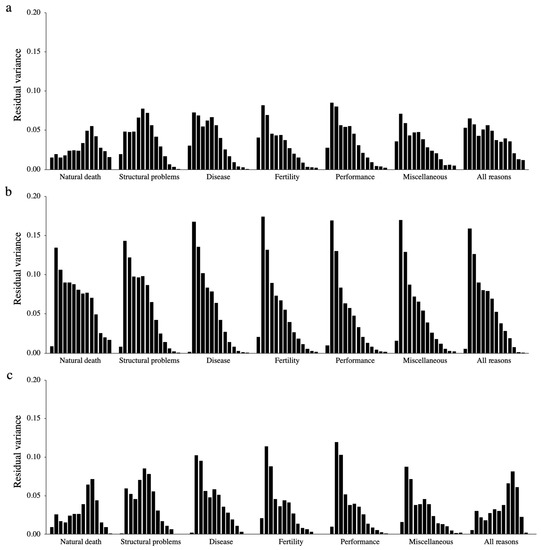
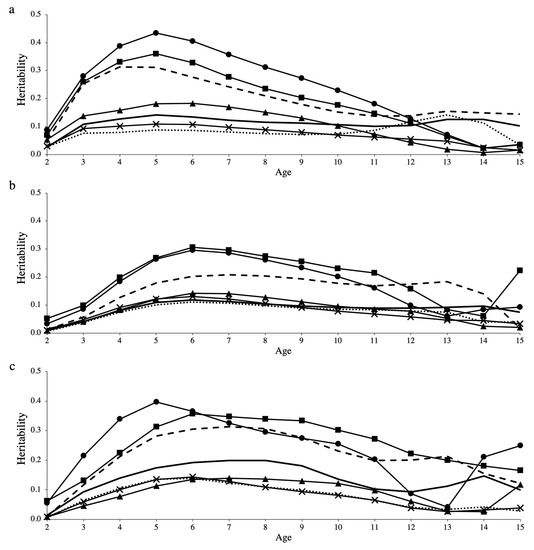
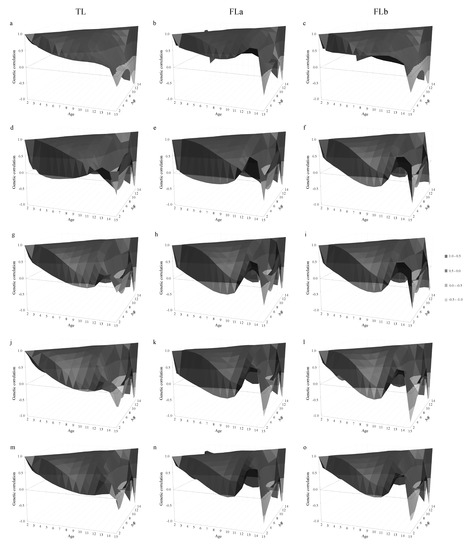
Heat stress severely affects dairy cattle production and reproduction performances in tropical regions. Genetic selection to maintain adequate yield and reproductive performance while enhancing their ability to withstand heat is essential for improving the genetics of dairy cows. Therefore, in this study, we aimed to estimate genetic parameters affecting production and reproduction performances under heat stress conditions in dairy cattle and to investigate the threshold point of heat stress for milk yield (MY), milk fat-to-protein ratio (FPR), and conception rate (CR) in Thai–Holstein dairy cattle. The data included 168,124 records related to MY and milk FPR and 21,278 records of CR in Thai–Holstein dairy cattle, covering the period from 1990 to 2007. A multiple-trait threshold-linear random regression model based on a Bayesian approach via Gibbs sampling was used to estimate variance components, genetic parameters (heritability values, and genetic correlations), and decline rates for each studied trait. The threshold point of heat stress was identified as a temperature and humidity index (THI) of 76. At THI76, a decline was observed in the MY, milk FPR, and CR of Thai dairy cattle. The heritability estimates for MY, milk FPR and CR were 0.347 ± 0.032, 0.293 ± 0.021, and 0.032 ± 0.001, respectively. The genetic correlation between MY and milk FPR and MY and CR were −0.24 and −0.53, respectively, whereas those between milk FPR and heat tolerance as well as between CR and heat tolerance were −0.48 and −0.49, respectively. In addition, the decline rates in MY, milk FPR, and CR were found to be associated with a high percentage of Holstein genetics. In conclusion, the results obtained in this study reveal that the simultaneous consideration of the MY, milk FPR, CR, and heat tolerance traits of Thai–Holstein dairy cattle is possible. In addition, developing a genetic model that incorporates THI is essential for sustainably addressing heat stress problems.
Full article

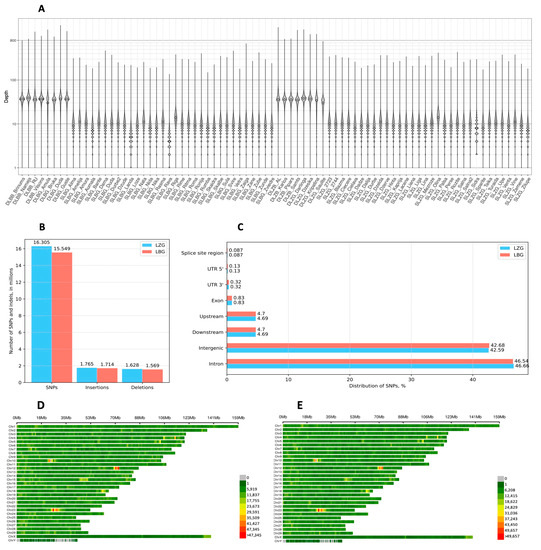
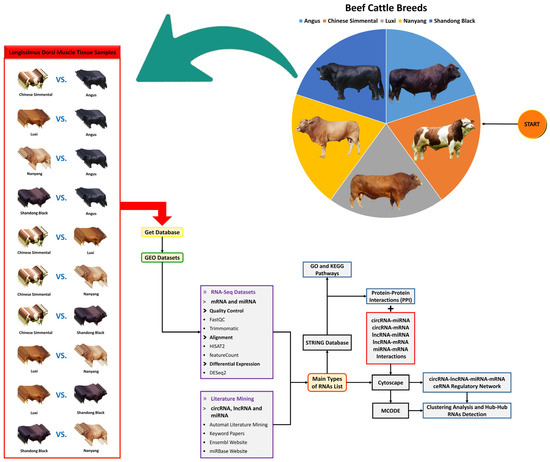
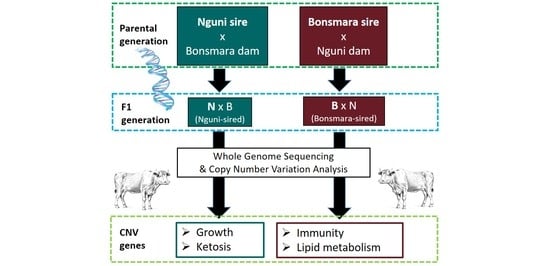
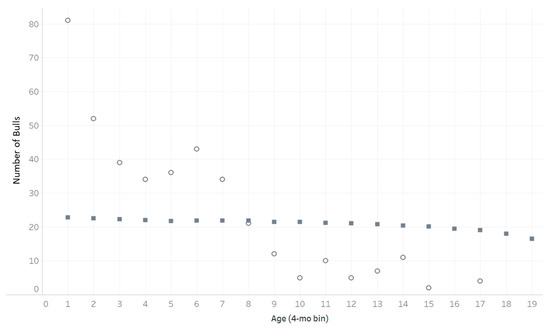
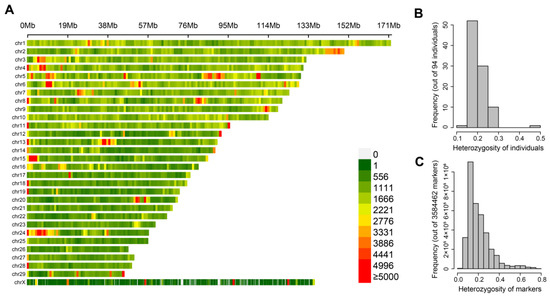
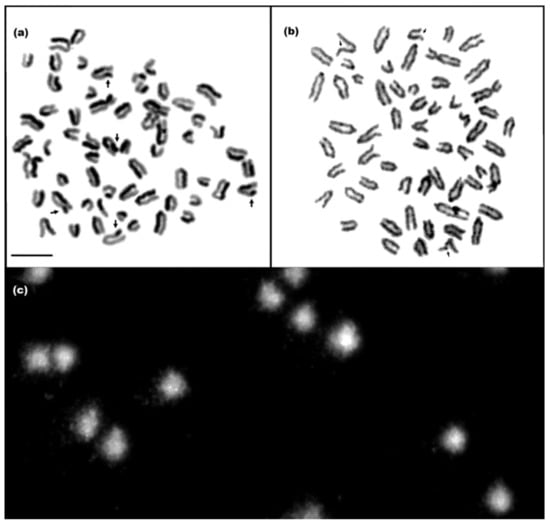
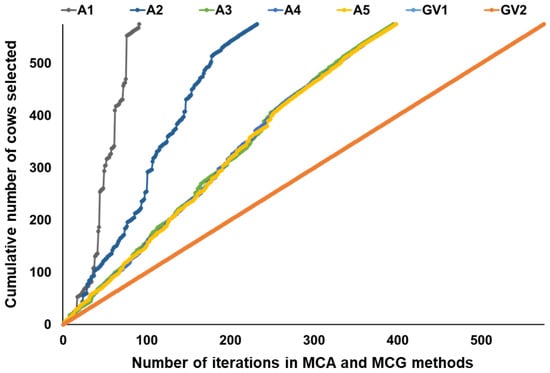
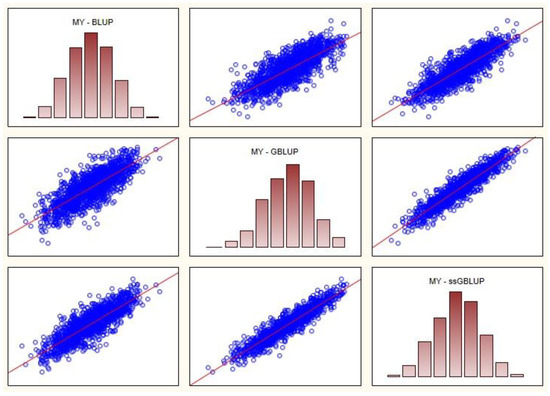
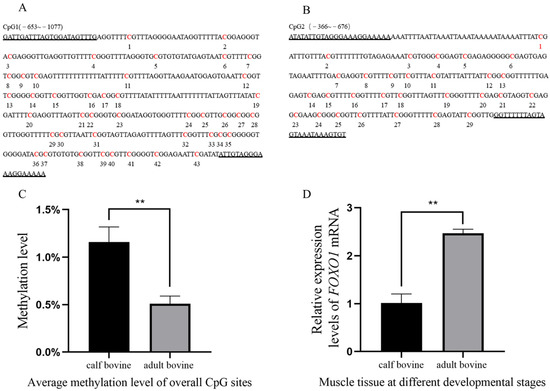
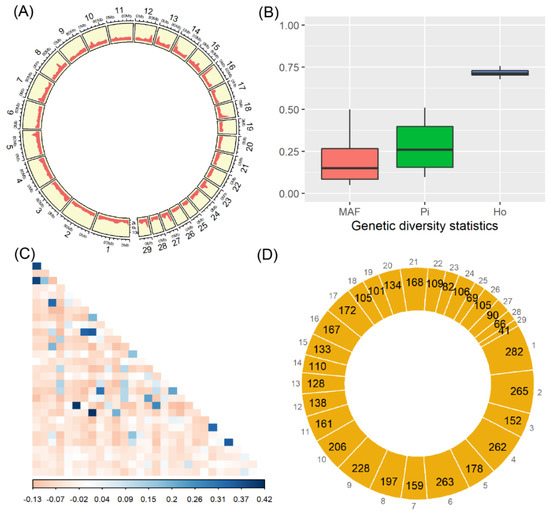
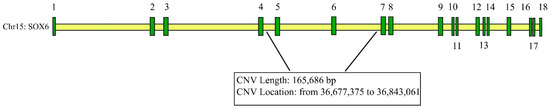
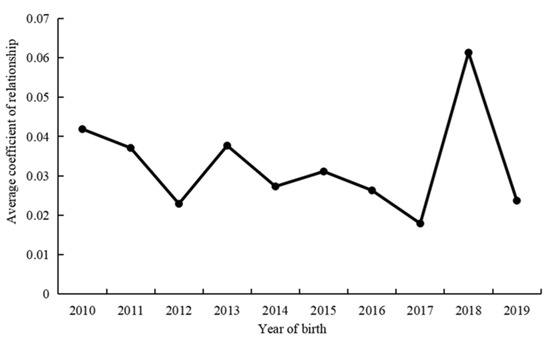
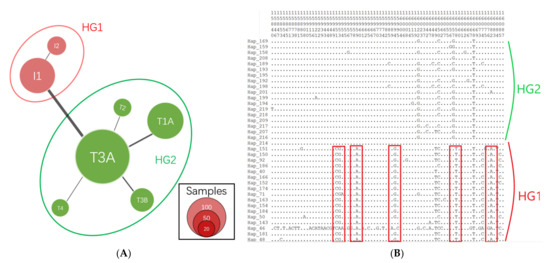
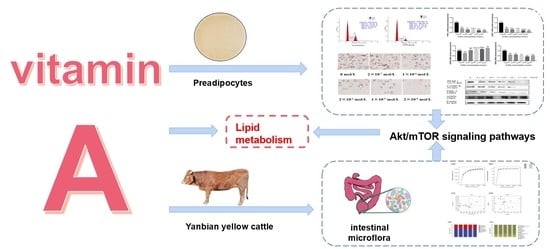
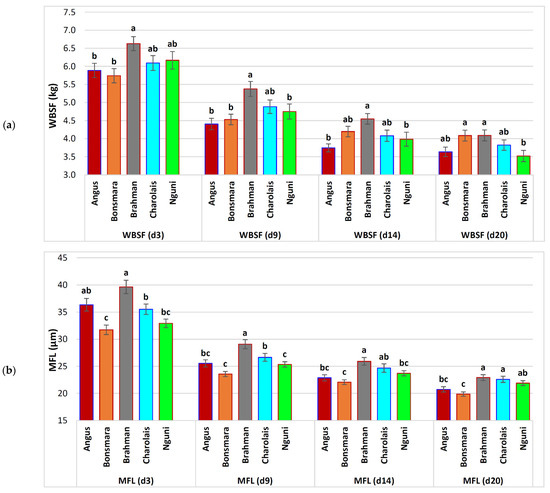
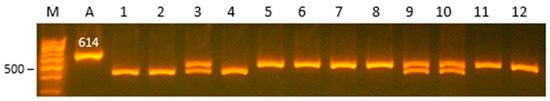
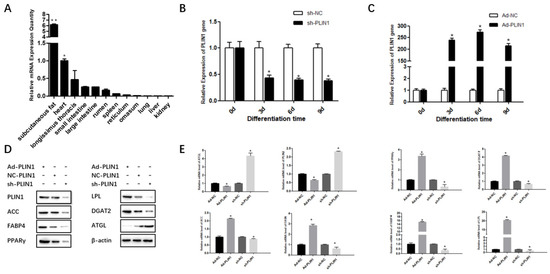
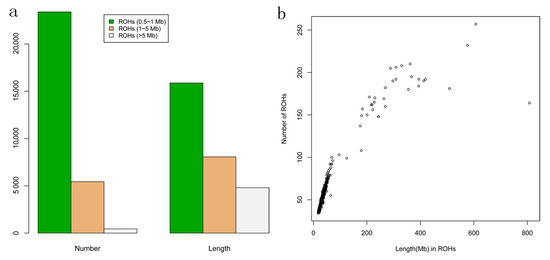
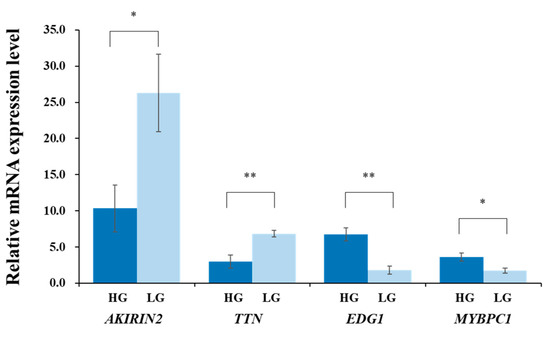
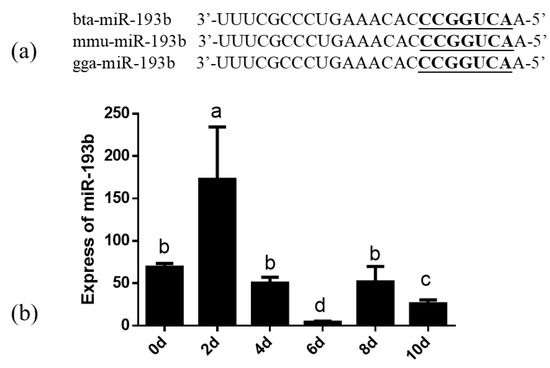
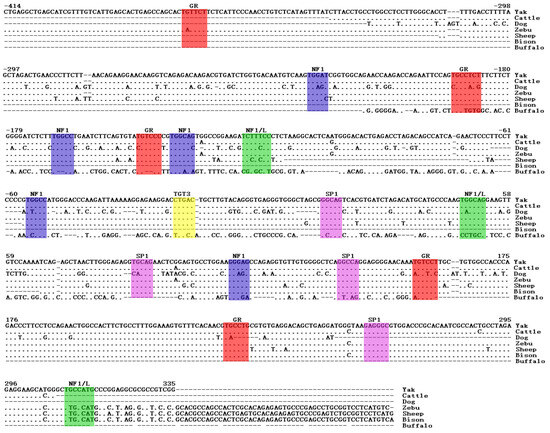
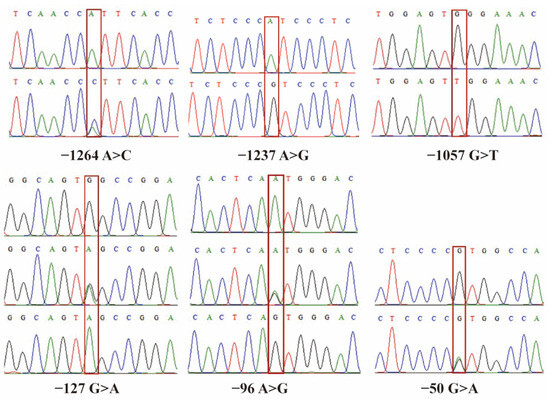
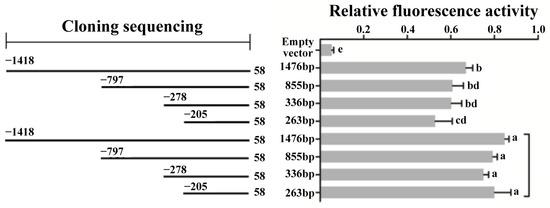
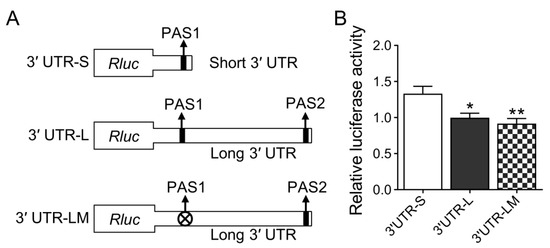
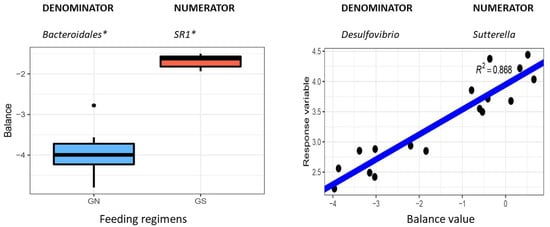
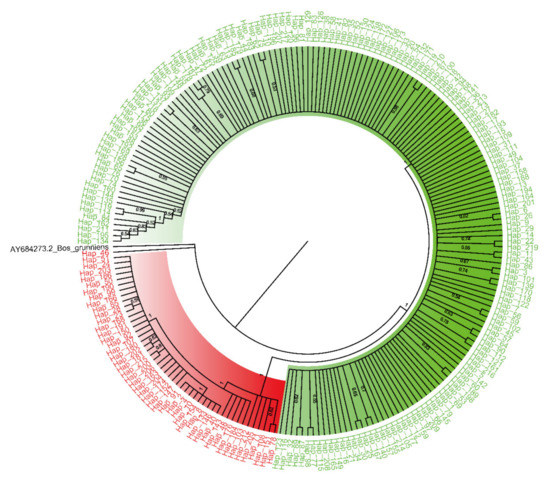
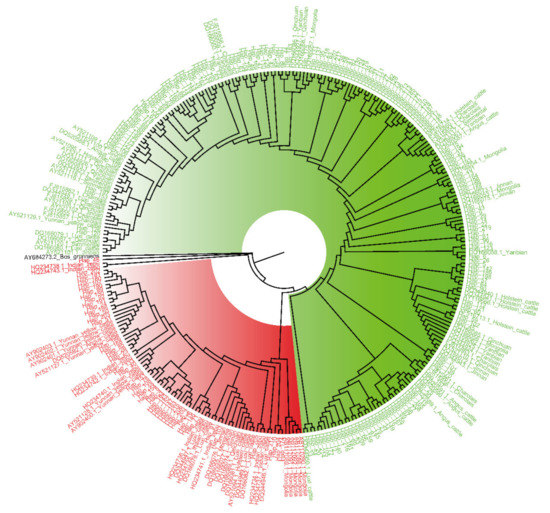
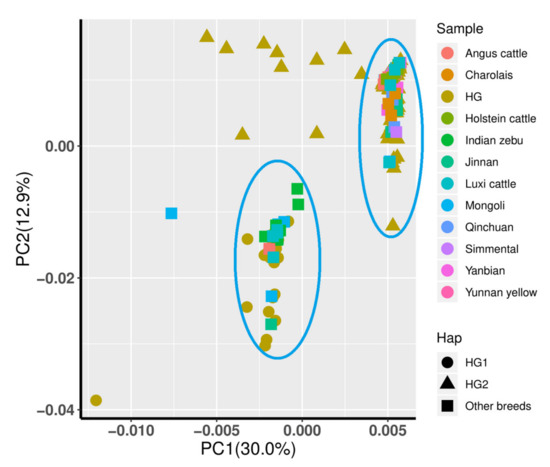
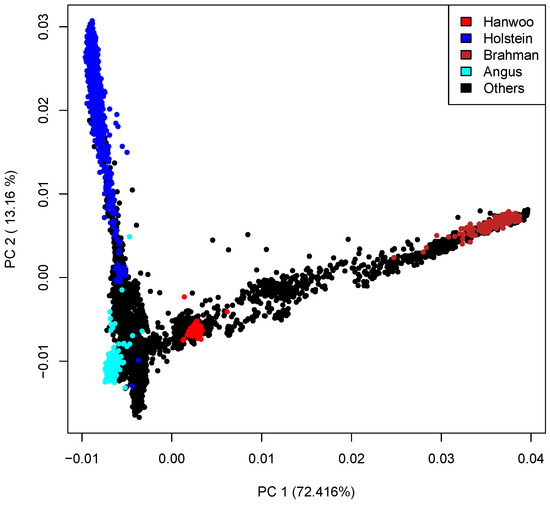
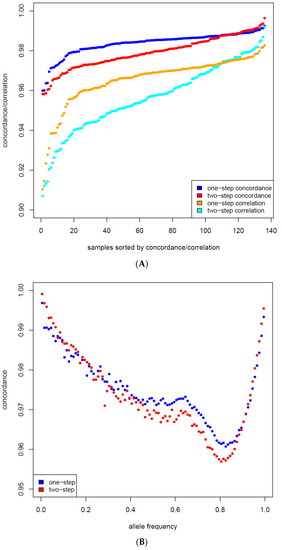
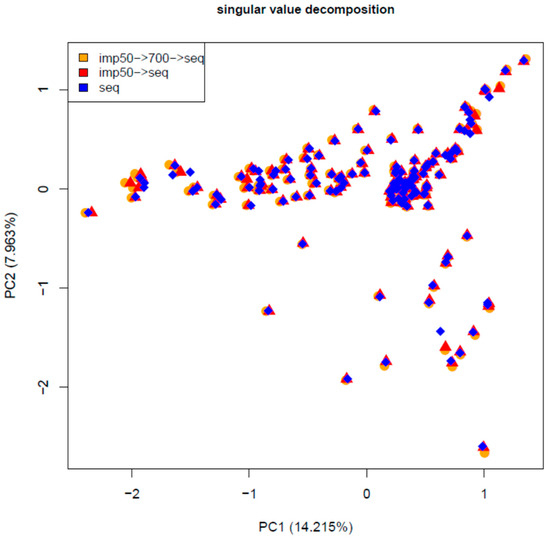
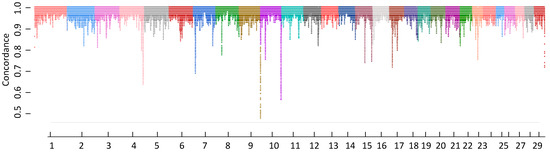
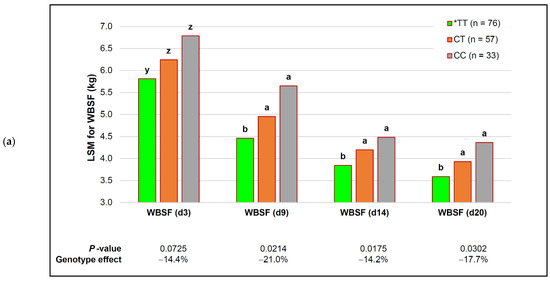
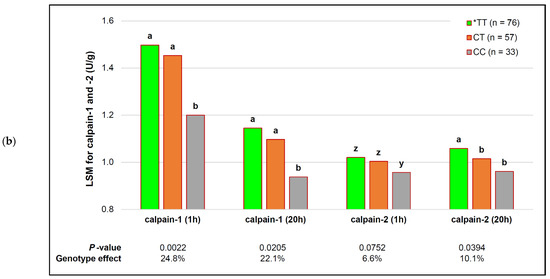
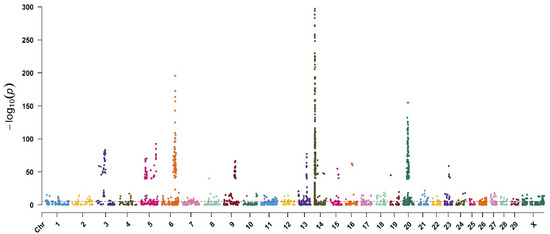
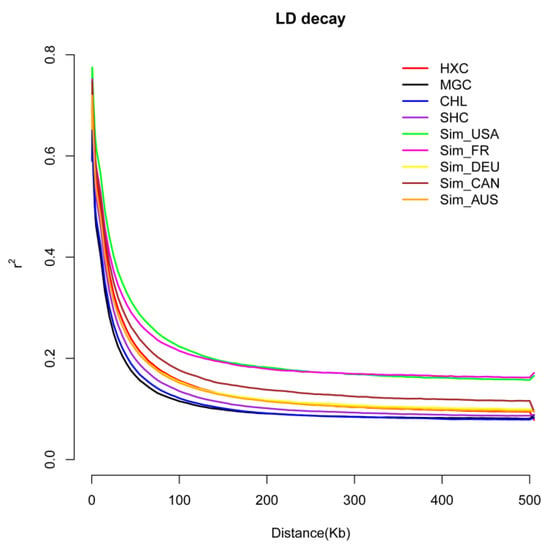
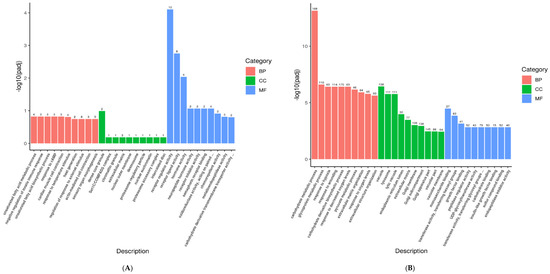
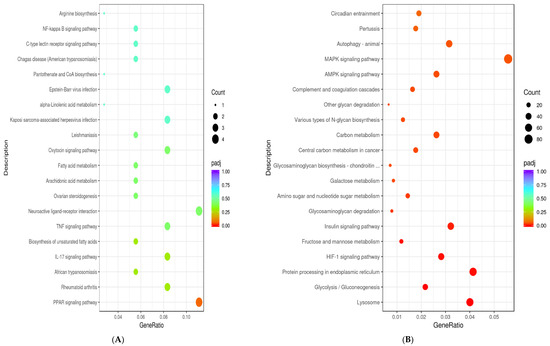
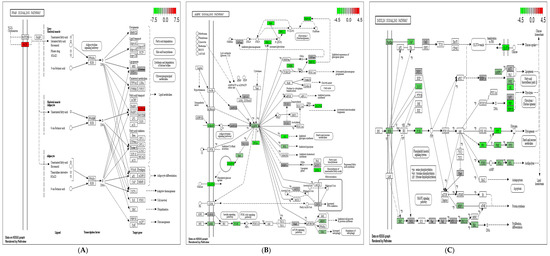
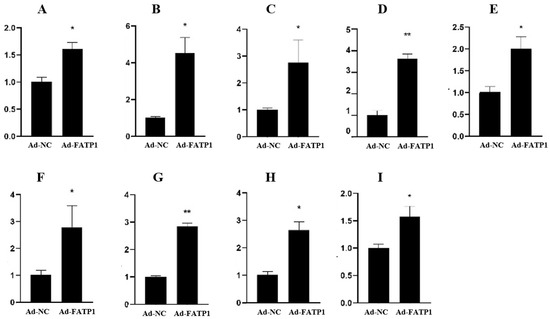
Figure 1



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}