Abstract
Gestational trophoblastic diseases (GTDs) encompass a spectrum of conditions characterized by abnormal trophoblastic cell growth, ranging from benign molar pregnancies to malignant trophoblastic neoplasms. This systematic review explores the molecular underpinnings of GTDs, focusing on genetic and epigenetic factors that influence disease progression and clinical outcomes. Based on 71 studies identified through systematic search and selection criteria, key findings include dysregulations in tumor suppressor genes such as p53, aberrant apoptotic pathways involving BCL-2 (B-cell lymphoma), and altered expression of growth factor receptors and microRNAs (micro-ribose nucleic acid). These molecular alterations not only differentiate molar pregnancies from normal placental development but also contribute to their clinical behavior, from benign moles to potentially malignant forms. The review synthesizes insights from immunohistochemical studies and molecular analyses to provide a comprehensive understanding of GTD pathogenesis and implications for personalized care strategies.
1. Introduction
Gestational trophoblastic diseases (GTDs) encompass a spectrum of conditions that range from benign to malignant forms and are characterized by abnormal growth of trophoblastic cells. The benign forms of GTD comprise molar pregnancies, which include complete hydatidiform moles (CHMs) and partial hydatidiform moles (PHMs). These two entities have varying clinical presentations and outcomes [1]. The malignant forms of GTD, also known as gestational trophoblastic neoplasms (GTNs), include choriocarcinoma (CC) and invasive moles (IMs), epithelioid trophoblastic tumors (ETTs), and placental-site trophoblastic tumors (PSTTs). These neoplasms can arise de novo after a normal pregnancy, or, most commonly, following a molar pregnancy [2] (Figure 1).
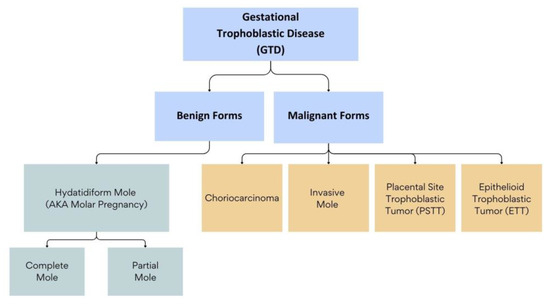
Figure 1.
Classification of Gestational Trophoblastic Diseases (GTDs).
Molar pregnancies typically result from errors in gamete formation and the fertilization process, which lead to the formation of an abnormal zygote with an abnormal amount of parental genetic material contributions. In CHM, the total amount of genetic material is normal, but all chromosomes are paternal without maternal chromosomes [2]. In CHM, an enucleated egg is either fertilized by one sperm, whose genetic material then duplicates (more common), or is fertilized by two sperms (less common). The result is a diploid zygote with 46 paternal chromosomes and no maternal genetic material (i.e., androgenetic diploid) [3]. However, there is a subtype of CHM called familial recurrent hydatidiform moles (FRHMs), in which cells are biparental diploid (i.e., cells contain 23 paternal chromosomes and 23 maternal chromosomes) [4,5]. This entity will be discussed in detail later in this review. In PHM, a normal egg (1n) is fertilized by more than one sperm, resulting in polyploidy [3]. Usually, there is triploidy, with an extra paternal haploid set of chromosomes, with the most common type being 69, XXX (90% of cases) [6].
The incidence of molar pregnancies varies across different world regions, ranging from 0.2 to 9.9 per 1000 pregnancies. However, its incidence is higher in Asian and African ethnicities [7]. The possibility of developing post-molar GTN is the primary reason for extended follow-up with serial beta-human chorionic gonadotropin (β-HCG) monitoring in patients after the evacuation of molar pregnancies. The incidence of post-molar pregnancy is around 15–20% in CHM but is only 0.5–5% in PHM [8,9]. This discrepancy is the reason follow-up protocols after the evacuation of a molar pregnancy differ for each subtype. Thus, it is essential to differentiate between the two entities during histopathological testing. The diagnosis of molar pregnancy follows specific morphological criteria through histopathological examination. The microscopic appearance differs between CHMs, PHMs, and hydropic abortions (HAs) [10]. However, challenges arise in differentiating between CHM and PHM, and even between molar pregnancy and HAs, due to overlapping histological signs, especially in the early stages of pregnancy [11,12]. The advent of ultrasonographic machines has made it possible to diagnose molar pregnancies in the early stages, which has paradoxically led to a more confusing morphological assessment and sometimes erroneous interpretation of tissue specimens during histopathological examination [11,12]. Multiple ancillary techniques utilizing immunohistochemical staining and genetic analyses have emerged to help differentiate between CHMs, PHMs, and HAs [13].
Research into these ancillary techniques has shed light on the intricate molecular mechanisms driving molar pregnancies. These include dysregulations in tumor suppressor genes such as p53, apoptotic pathways involving BCL-2 (B-cell lymphoma-2), and aberrant expression of growth factor receptors and microRNAs (micro-ribose nucleic acid) [14,15]. These molecular alterations not only differentiate molar pregnancies from normal placental development but also contribute to their clinical behavior, ranging from benign moles to potentially malignant forms [16,17,18]. Figure 2 summarizes the key molecular mechanisms described in this review. This review explores the current understanding of molecular markers and genetic mutations in molar pregnancies, emphasizing their roles in cell proliferation, apoptosis, and trophoblastic differentiation. By synthesizing findings from immunohistochemical studies and molecular analyses, this article aims to provide insights into the pathogenesis of GTDs.
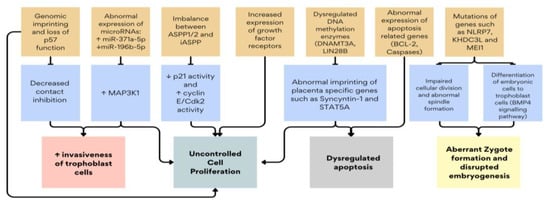
Figure 2.
Summary of the key molecular dysregulations in GTD. ↑: increased gene expression or protein/enzyme activity, ↓: decreased gene expression or protein/enzyme activity.
2. Materials and Methods
2.1. Search Strategy
A systematic search was conducted across electronic databases including PubMed, Scopus, and CINAHL EBSCO. Medical Subject Headings (MeSH) terms and free-text keywords such as “Hydatidiform mole”, “gestational trophoblastic disease”, and “molar pregnancy” were used in conjunction with “gene”, “genome”, “genetic”, “immunohistochemistry” or “molecular” in February 2024 to retrieve relevant data from January 2004 to January 2024. Additionally, the references for pertinent studies were manually searched if they were not included in these databases.
2.2. Eligibility Criteria
2.2.1. Inclusion Criteria
Studied were included if they met all the following criteria—1, 2, and 3:
- Articles written in the English language or an English translation is available;
- Full-text articles reporting on human genes, genome, genetics, or molecular bases;
- Articles containing information on gestational trophoblastic disease, hydatidiform mole, or molar pregnancy.
All eligible studies published between January 2014 and January 2024 were included for review. No retracted papers were included in this review.
2.2.2. Exclusion Criteria
Exclusion criteria included duplicate studies, review articles, non-genetic and non-immunohistochemical studies, articles not available in the English language, and studies where full-text articles were only available with payment. Conference abstracts, expert opinions, and critical appraisals were also excluded from the review.
2.3. Study Selection
After a comprehensive search of the databases, a total of 5259 results were initially retrieved. All the abstracts and study titles were screened, and duplicates were removed. Furthermore, there were a total of 5188 studies excluded, as they either did not fit the inclusion criteria, were animal studies, included only gestational trophoblastic neoplasia, placental abnormalities other than hydatidiform mole, or did not explore the genetic basis of the disease. Eventually, 71 articles met all the criteria and were included in the review. A summary of the studies included in this review is available as Supplementary Material Table S1.
2.4. Data Collection
All authors (SB, RD, MA, MG, RS, RK, and SSK) independently reviewed all titles. The potential relevance of studies for inclusion in the review was determined through discussion. Selected titles and abstracts were further screened to avoid overlap of cases. Full-text copies of the selected papers were obtained, and the same reviewers independently extracted relevant data regarding study characteristics, results of molecular testing, and significant associations with GTD outcomes. Whenever possible, single case reports were cross-checked with other reports from the same location and hospital. Finally, studies were screened by assessing their suitability for inclusion in the evidence acquisition for the molecular basis of hydatidiform mole. Figure 3 illustrates the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA).
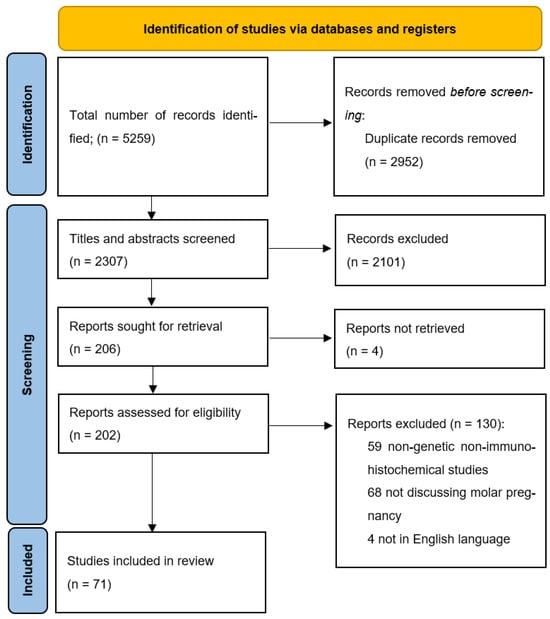
Figure 3.
PRISMA flow diagram for study inclusions.
3. Results and Discussion
3.1. Genomic Imprinting
Genomic imprinting, regulated by DNA (deoxyribose nucleic acid) methylation at differentially methylated regions (DMRs), governs the expression of genes based on their parental origin [19]. In complete moles, which are predominantly androgenetic diploids, the absence of maternal genomic contribution results in significant implications for imprinted genes such as p57kip2 (p57 cyclin-dependent kinase inhibitor 2) [5,6,20]. P57kip2, paternally imprinted and expressed exclusively from the maternal allele, exhibits markedly reduced expression in CHMs compared to partial moles and normal placentas [21,22,23,24,25,26,27]. This distinct immunohistochemical pattern of p57kip2 has become crucial for distinguishing between different types of hydatidiform moles [11,12,28]. Studies using human trophoblast stem cells have highlighted p57kip2’s role in regulating contact inhibition, suggesting that its diminished expression contributes to the uncontrolled proliferation characteristic of CHMs [29]. Despite rare instances of biparental diploid CHMs [5,30] and retained maternal copy of respective chromosomes [31], the majority exhibit abnormal genomic imprinting and epigenetic dysregulation, including down-regulation of DNMT3A (DNA Methyltransferase 3 Alpha) and LIN28B (Lin-28 Homolog B), key enzymes for DNA methylation and parental imprinting maintenance [32,33,34]. Sanchez-Delgado et al. confirmed abnormalities in the epigenomic regulation of placenta-specific maternally inherited genes, characterized by atypical methylation patterns [35]. Abnormal methylation patterns affect genes like ERVWE1 (Syncyntin-1), implicated in syncytiotrophoblast function and apoptosis regulation, further underlining the impact of epigenetic changes in CHM pathogenesis [36,37]. Abnormal methylation affects other genomic components, including LINE-1 (Long Interspersed Nuclear Element-1), a transposable element, and the gene STAT5A [38,39]. While typically p57-negative, rare cases of CHM show aberrant p57 expression linked to retained maternal chromosomes or biparental diploidy, emphasizing the utility of genotyping in diagnosing and understanding the molecular basis of molar pregnancies [40,41,42,43]. Similarly, although partial hydatidiform moles are typically p57-positive, rare instances (1.3%) have been reported with negative p57 results [44]. Genomic imprinting in gametogenesis is responsible for genetic expression in the offspring depending on parent-of-origin, and the human placenta has a high and prolific expression of imprinted genes [45,46]. The genotype of molar pregnancies, particularly heterozygous/dispermic complete moles, correlates with a higher risk of developing post-molar gestational trophoblastic neoplasia, suggesting a potential role for additional paternal imprints in promoting trophoblastic proliferation [16]. Further research is needed to elucidate the precise mechanisms underlying these genomic imprinting abnormalities and their clinical implications in molar pregnancy pathogenesis [1,2,3,4].
3.2. Recurrent Hydatidiform Mole
Recurrent hydatidiform moles (RHMs) encompass two distinct categories: sporadic cases and familial forms. Sporadic RHM typically occurs with a recurrence rate of 1–6% among individuals with a history of molar pregnancy [47]. These cases often exhibit a monospermic or dispermic androgenetic diploid genotype [5]. In contrast, cases of familial RHM (FRHM) are generally biparental diploids and characterized by two or more affected female family members experiencing recurrent molar pregnancies [4,5]. These familial cases are considered autosomal recessive inherited disorders and are associated with abnormal CpG methylation in imprinted genes [48].
The predominant genetic culprits linked to FRHM include mutations in NLRP7 (Nucleotide-Binding Oligomerization Domain, Leucine Rich Repeat and Pyrin Domain-Containing 7) and KHDC3L (KH Domain Containing 3 Like), with NLRP7 mutations found in 40–80% and KHDC3L mutations in 10–14% of affected individuals [47,49]. A mutation analysis by Nguyen et al. of 113 patients with recurrent molar pregnancies showed that mutations of NLRP7 and KHDC3L were associated with diploid biparental HM, while recurrent molar pregnancies without mutations were associated mostly with diploid androgenic monospermic and triploid biparental dispermic [50].
Multiple case reports and case series identified mutations in the homozygous state or combined heterozygous states involving the NLRP7 and KHDC3L genes associated with recurrent hydatidiform moles [48,51,52,53,54,55,56]. NLRP7, located on chromosome 19q13.4, is a maternal effect gene, which means that the phenotype is influenced by mutations in maternal genes only, crucial for oocyte and embryo development [57]. It localizes in the cytoskeleton, which contains microtubules essential for cellular division [57]. Homozygous mutations in NLRP7 disrupt embryo development by impairing cellular division and organization within the cytoskeleton, leading to developmental arrest and failed embryo progression. In a study investigating 10 embryos from a woman with recurrent hydatidiform mole and a homozygous pathogenic variant in NLRP7 (c.2810+2T>G), all embryos exhibited developmental arrest and did not advance to a stage suitable for transfer [58]. This mutation primarily affects embryos when inherited maternally, owing to its maternal inheritance pattern [48]. Additionally, NLRP7 interacts with YY1 (Yin Yang 1) to activate the BMP4 (Bone Morphogenetic Protein 4) signaling pathway, stimulating embryonic cells to differentiate into trophoblasts rather than maintaining pluripotency to differentiate into various cell types, thereby contributing to mole formation [49,59]
KHDC3L, though less frequently implicated in biparental complete hydatidiform mole (BICHM), likely forms complexes with NLRP7 to assist in oogenesis and early embryonic development [39]. Mutations in these genes can dysregulate inflammatory cytokine secretion within the endometrial cavity, potentially altering the implantation microenvironment and contributing to molar pregnancy pathogenesis [39]. Specifically, mutations in NLRP7 have been associated with lower levels of immune signaling molecules like IL-1β [60]. The altered immune function observed in patients with NLRP7 mutations may be relevant to understanding the pathogenesis of molar pregnancy. Reddy et al. identified 11 NLRP7 variants in homozygous or compound heterozygous state in individuals with a history of recurrent hydatidiform mole. Some of these variants were associated with abnormalities in transcription and with post-transcriptional modifications of mRNA (messenger ribose nucleic acid) that affect splicing and result in either the absence of gene transcription or the formation of abnormally long or truncated proteins [61]. Reddy et al.-identified NLRP7 variants along with other variants associated with RHM are presented in Table 1 [47,48,52,53,54,55,56,58,61,62,63,64,65,66,67,68].

Table 1.
Genetic variants and mutations associated with Recurrent Hydatidiform Mole (RHM).
NM et al. examined the interaction of the p57KIP2 gene with NLRP7 mutations in hydatidiform mole (HM). Analyzing 36 products of conception (POCs) from patients with two defective NLRP7 alleles revealed that all samples were diploid biparental [69]. They found that the expression of p57KIP2 varied based on the type of NLRP7 mutation: missense mutations were associated with positive p57KIP2 expression and features similar to partial HMs, while protein-truncating mutations correlated with negative p57KIP2 expression and characteristics of complete HMs [69]. These results suggest that NLRP7 mutations impact trophoblast cell differentiation and proliferation to varying degrees, with severe mutations leading to excessive proliferation and mild mutations allowing some differentiation.
In addition to NLRP7 and KHDC3L, other gene variants have been identified in RHM cases, such as MEI1 (meiotic double-stranded break formation protein 1), TOP6BL/C11orf80 (type 2 DNA topoisomerase 6 subunit B-like), and REC114 (Recombination Protein 114), all of which affect oocyte meiosis and early embryonic development [67]. These mutations can result in abnormal spindle morphology, misaligned chromosomes, extrusion of all chromosomes into the polar body, and other defects that can lead to empty oocytes, androgenetic zygote formation, and subsequent molar pregnancy [67].
Understanding the genetic underpinning of diseases through comprehensive mutation analysis provides insights into the complex mechanisms involved [70] and has the potential to offer avenues for personalized care in affected individuals. Similarly, the genetic and molecular basis of different molar pregnancies has a pivotal role in decisions about treatment protocols and follow-ups.
3.3. Molecular Dysregulations in GTD
Immunohistochemical studies on hydatidiform moles offer valuable insights into the molecular underpinnings that distinguish them from normal placentas and hydropic abortions. These investigations reveal significant alterations in markers crucial for cell cycle regulation, apoptosis, and trophoblast differentiation, shedding light on the pathogenesis of molar pregnancies. Table 2 summarizes the molecular dysregulations associated with Gestational Trophoblastic Disease [14,68,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85].
Table 2.
Molecular dysregulation in GTD.
The expression of p53 in cytotrophoblasts is markedly elevated in molar pregnancies compared to hydropic abortions [14,80,81]. Studies indicate higher levels of p53 expression in patients with invasive moles and choriocarcinoma compared to those with non-invasive hydatidiform moles [86,87,88]. Hadi et al. demonstrated that more than 55% positive staining for the TP53 gene can effectively distinguish non-invasive hydatidiform mole from invasive forms and choriocarcinoma with 100% sensitivity and 92.9% specificity, albeit based on a small sample size [88].
P53, recognized as a tumor suppressor gene and often referred to as the “guardian of the genome”, is normally expressed in cytotrophoblasts and infrequently in stromal cells [81]. Its primary functions include inducing cell cycle arrest and apoptosis [81,89]. This is achieved through the transcriptional activation of p21/WAF1, which interacts with cyclin E/Cdk2 and cyclin D/Cdk4 complexes, leading to G1 arrest in the cell cycle [89]. Therefore, p53 serves as a marker of proliferative activity, crucial for regulating proliferation by inducing either cell cycle arrest or apoptosis. The overexpression of p53 in molar tissues reflects the heightened proliferative capacity of trophoblastic cells [81]. Similarly, Studies have demonstrated significantly higher expression of p63, a tumor suppressor gene from the p53 family, in molar pregnancies compared to hydropic abortions (p-value < 0.05) [72,83].
The ASPP (Ankyrin-repeat, SH3-domain, and proline-rich region containing protein) family, including ASPP1 and ASPP2, modulates p53 activity and is found to be dysregulated in gestational trophoblastic diseases (GTD) [90,91]. Normally functioning as tumor suppressors, ASPP1 and ASPP2 stimulate p53-mediated transcriptional modifications of p21. However, Mak et al. have reported their downregulation in GTD [90,91]. Another crucial member of this family is iASPP (inhibitory ASPP), which exhibits increased expression in complete hydatidiform mole and choriocarcinoma compared to normal placental tissue [92]. Chan et al. demonstrated that silencing iASPP was associated with reduced production of autophagy-related proteins (LC3) and increased susceptibility to oxidative stress in choriocarcinoma cells [92]. These findings underscore the role of imbalanced expression of ASPP1/2 (downregulation) and iASPP (upregulation) in the pathogenesis of GTD [90,91,92].
Several mutations in p53 genes have also been identified in patients with molar pregnancy. Chan et al. identified two missense mutations (p.R249S and p.R248Q) that disrupt p53 DNA binding sites, impairing its ability to control cell proliferation [93]. Additionally, a nonsense mutation (p.R213X) was reported to prematurely truncate the protein, resulting in loss of its normal function [93]. Several studies showed that Ki-67 (Kiel-67) exhibits heightened expression levels in hydatidiform moles compared to both hydropic abortions and normal placental tissues [72,77]. This protein plays a crucial role in trophoblast differentiation and is primarily expressed by cytotrophoblasts. Notably, studies have shown significant overexpression of Ki-67 in CHM when analyzed in conjunction with Cyclin E, a key promoter of cell cycle progression, demonstrating a statistically significant difference compared to normal and hydropic placentas (p-value < 0.05) [94].
Apart from aberrant cell proliferation, dysregulation of apoptosis also plays a pivotal role in the pathogenesis of GTD. Studies examining the apoptotic index in molar pregnancies yield disparate findings, primarily due to methodological variations in apoptosis assessment. One study focused on caspase-3, a critical enzyme in caspase-dependent apoptosis, and reported diminished expression levels in GTN, suggesting reduced apoptosis [95]. In contrast, another study utilizing the TUNEL assay (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling), which detects DNA fragmentation as a late-stage apoptosis marker, indicated heightened apoptotic activity in GTD [96]. Furthermore, a study comparing the expression of caspases between persistent CHM and regressed CHM found no significant difference in apoptosis levels, though it was limited by a small sample size [97]. These findings suggest that while apoptosis levels are elevated in GTD, it may predominantly occur through caspase-independent pathways rather than the traditional caspase-dependent pathway. This underscores the necessity for further research to elucidate the specific apoptotic mechanisms at play in molar pregnancies and their clinical implications.
BCL-2, an anti-apoptotic gene, plays a crucial role in regulating caspase-dependent apoptosis, and acts as an anti-proliferative protein by inhibiting cell transition from quiescence to the S-phase [98]. In the placenta, BCL-2 expression in syncytiotrophoblasts controls apoptosis in these multinuclear cells. It prevents the spread of apoptotic changes and the fragmentation of other nuclei sharing the same cytoplasm when apoptosis occurs in one nucleus of the syncytiotrophoblast [96]. Studies assessing BCL-2 expression in molar pregnancies have yielded conflicting results. Several studies have demonstrated significantly reduced BCL-2 immunohistochemical staining in the syncytiotrophoblasts of complete mole compared to control specimens [68,71,73,99]. Lin et al. demonstrated that CHM progressing to gestational trophoblastic neoplasia expressed higher levels of miR-181b-5p and miR181d-5p and lower levels of their target, BCL-2, compared to those that regressed following evacuation [100]. Conversely, other studies have reported increased BCL-2 expression in molar pregnancies or found insignificant staining differences between molar and non-molar placentas [72,101,102]. Missaoui et al., in their evaluation of 220 specimens classified based on morphological appearance and molecular markers into 140 CHMs, 41 PHMs, and 39 HAs, found significantly higher BCL-2 immunostaining in partial mole (61%) and CHM (73.6%) compared to hydropic abortions (7.7%, p = 0.001, p < 0.0001 respectively), which may explain earlier findings of reduced caspase expression in GTD [87,89]. Larger studies incorporating reliable diagnostic criteria and ploidy analysis are warranted to accurately delineate the alterations in BCL-2 expression in molar pregnancy. In contrast, the immunoexpression of Bax, another regulator gene in the apoptosis pathway, did not show significant differences between molar pregnancies and non-molar pregnancies according to Reza et al. [71].
The molecular distinctions between different types of GTDs are crucial in identifying markers for the behavior and aggressiveness of molar pregnancies. Research has identified distinct patterns of growth factor receptor expression in various trophoblast types. Specifically, EGFR ( Epidermal Growth Factor Receptor) and ERBB4 (Erythroblastic Leukemia Viral Oncogene Homolog 4) are notably overexpressed in actively proliferating trophoblasts [16]. Additionally, trophoblasts exhibiting invasive characteristics demonstrate heightened expression of ERBB2, also known as HER2/neu (Human Epidermal Growth Factor Receptor 2) and C-erbB-2 (Cellular Erythroblastic Leukemia Viral Oncogene Homolog 2), and ERBB3 [103,104]. Epidermal Growth Factor (EGF) and Heparin-Binding EGF-like Growth Factor (HB-EGF) enhance cell signaling pathways in EGFR-expressing trophoblasts, thereby promoting cell cycle progression and contributing to proliferative activity in hydatidiform moles [16].
C-erbB-2 was also found by Erol et al. to be significantly overexpressed in complete moles compared to partial moles and hydropic abortions (HAs) [73]. Previous studies suggest that this overexpression may correlate with aggressive behavior in CHM [17,18]. Another tyrosine receptor kinase, CD117 (c-KIT), presents on various cell types including mast cells, hematopoietic stem cells, and germ cells [105], exhibits increased activation in molar tissues compared to hydropic abortions, and is associated with aggressive CHM behavior [73]. Activation of CD117 through stem cell factor (SCF) regulates processes such as proliferation, cell differentiation, apoptosis, and cell adhesion [105,106].
miRNAs have been implicated in GTDs based on their distinct expression profiles in trophoblastic tissues. Among these, miR-371a-5p is notable for its oncogenic properties in various cancers and is found to be upregulated in progressive CHMs, suggesting a potential role in the progression of malignancy [15]. Another miRNA, miR-196b-5p (miR-196b), functions as a tumor suppressor and exhibits reduced expression in CHM tissues compared to normal placentas [78]. This decrease correlates with elevated levels of Mitogen-activated protein kinase 1 (MAP3K1), a protein promoting cell proliferation and differentiation [78]. Increasing miR-196b expression in hydatidiform mole cells has been shown to diminish their proliferation and invasion, suggesting its potential as both a diagnostic marker and therapeutic target [78]. Furthermore, miR-21 is significantly overexpressed in tissues from hydatidiform moles, where it enhances aggressive behaviors such as proliferation, migration, and invasion of trophoblastic cells. This characteristic makes miR-21 a promising candidate for targeted approaches in the diagnosis and treatment of gestational trophoblastic neoplasms [79].
Twist-1, a negative regulator of E-cadherin, shows significantly elevated levels in CHM and correlates with disease invasiveness [75,84,85]. E-cadherin, crucial for cell-to-cell adhesion, exhibits reduced expression associated with GTD and is associated with increased invasiveness [14,74,75,84], although one study with a small sample failed to show a significant association [74].
Thiol group-containing compounds, such as glutathione, are ROS scavengers. The thiol group (–SH) can donate a hydrogen atom in redox reactions to reduce oxidative stress, which results in the formation of disulfide bonds (–S–S–) [107]. Thus, a decreased thiol/disulfide ratio indicates a higher oxidative stress state. Peckan et al. demonstrated a decreased thiol/disulfide ratio in molar pregnancy compared to non-molar placental tissue, indicating higher oxidative stress in molar pregnancy. Similarly, Incebiyik et al. showed that markers for both oxidative stress and apoptosis (M30) were higher in CHM than normal pregnancy and parallel to each other [108]. Oxidative stress is generally higher during pregnancy and can be even more pronounced in pregnancy complications such as pre-eclampsia [109]. An imbalance between antioxidant capacity and reactive oxygen species (ROS) can lead to oxidative damage in the maternal and placental compartments [109].
Since the 1980s, numerous studies have demonstrated that GTD, particularly choriocarcinoma, and pre-eclampsia share common pathogenic mechanisms, including disruptions in angiogenesis, increased oxidative stress, and complement dysregulation [109,110,111,112]. Both pre-eclampsia and choriocarcinoma exhibit abnormalities in angiogenesis regulation. In pre-eclampsia, there are elevated levels of the antiangiogenic protein sFlt-1 (Soluble Fms-like Tyrosine Kinase 1) and reduced levels of angiogenic factors such as vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) [110]. Conversely, choriocarcinoma is associated with increased levels of the pro-angiogenic factor PlGF [110]. Given these parallels, mechanisms proven to be involved in pre-eclampsia warrant investigation in GTD. For instance, CD93, an angiogenic factor known to play a significant role in pre-eclampsia, has yet to be studied in the context of molar pregnancies [113]. Future research exploring the role of CD93 in GTD could reveal new potential therapeutic targets.
4. Conclusions
In conclusion, the molecular landscape of molar pregnancies unveils a complex interplay of genetic and epigenetic factors that influence disease progression and clinical outcomes. The overexpression of p53 in cytotrophoblasts and dysregulation of its modulators, ASPP1/2 and iASPP, highlights the proliferative potential and invasive nature of molar tissues. Moreover, discrepancies in proteins involved in apoptotic pathways such as BCL-2 and caspases highlight the dysregulation of the balance between cell survival and death in GTDs. The differential expression of growth factor receptors such as EGFR, ERBB2, and CD117 in trophoblasts reflects their roles in promoting cell proliferation and invasion, contributing to the aggressive behavior observed in some molar pregnancies.
Despite these advancements, challenges remain in standardizing diagnostic criteria and therapeutic approaches for molar pregnancies. Future research should focus on elucidating the specific molecular pathways driving molar pregnancy pathogenesis and developing targeted interventions to improve clinical outcomes. It may be specifically helpful in preventing the recurrence of GTD and GTNs. By integrating molecular insights with clinical observations, we can advance our understanding of GTDs and enhance patient care through personalized medicine strategies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25168739/s1.
Author Contributions
Conceptualization—R.D., R.K. and S.N.M.B.; methodology, R.D., S.N.M.B. and S.S.K.; software, S.N.M.B., M.G.B.K. and M.A.M.F.P.; validation, S.N.M.B., R.D. and R.A.A.S.; formal analysis, S.N.M.B.; investigation, S.N.M.B., M.A.M.F.P., M.G.B.K., S.S.K., R.A.A.S., R.K. and R.D.; resources, S.N.M.B., R.D., R.K., M.A.M.F.P., M.G.B.K. and S.S.K.; data curation, R.D. and S.N.M.B.; writing—original draft preparation, S.N.M.B. and R.D.; writing—review and editing, R.D., R.K. and R.A.A.S.; visualization, S.N.M.B. and R.D.; supervision, R.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lurain, J.R. Gestational trophoblastic disease I: Epidemiology, pathology, clinical presentation and diagnosis of gestational trophoblastic disease, and management of hydatidiform mole. Am. J. Obstet. Gynecol. 2010, 203, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.; Sorosky, J. Gestational Trophoblastic Disease; StatPearls: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470267/ (accessed on 26 June 2024).
- Ning, F.; Hou, H.; Morse, A.N.; Lash, G.E. Understanding and management of gestational trophoblastic disease. F1000Research 2019, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Moss, J.; Sebire, N.; Cui, Q.; Seckl, M.; Xiang, Y.; Fisher, R. Analysis of the chromosomal region 19q13.4 in two Chinese families with recurrent hydatidiform mole. Hum. Reprod. 2006, 21, 536–541. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kalogiannidis, I.; Kalinderi, K.; Kalinderis, M.; Miliaras, D.; Tarlatzis, B.; Athanasiadis, A. Recurrent complete hydatidiform mole: Where we are, is there a safe gestational horizon? Opinion and mini-review. J. Assist. Reprod. Genet. 2018, 35, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, V.A.; Butera, D.; Armano, G.; Monfardini, L.; Gaiano, M.; Gambino, G.; Sozzi, G.; Merisio, C.; Berretta, R. Obstetrics outcomes after complete and partial molar pregnancy: Review of the literature and meta-analysis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2021, 259, 18–25. [Google Scholar] [CrossRef]
- Tantengco, O.A.G.; De Jesus, F.C.C.; Gampoy, E.F.S.; Ornos, E.D.B.; Vidal, M.S.; Cagayan, M.S.F.S. Molar pregnancy in the last 50 years: A bibliometric analysis of global research output. Placenta 2021, 112, 54–61. [Google Scholar] [CrossRef]
- Ngan, H.Y.; Seckl, M.J.; Berkowitz, R.S.; Xiang, Y.; Golfier, F.; Sekharan, P.K.; Lurain, J.R.; Massuger, L. Diagnosis and management of gestational trophoblastic disease: 2021 update. Int. J. Gynecol. Obstet. 2021, 155, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Albright, B.B.; Shorter, J.M.; Mastroyannis, S.A.; Ko, E.M.M.; Schreiber, C.A.; Sonalkar, S. Gestational Trophoblastic Neoplasia after Human Chorionic Gonadotropin Normalization Following Molar Pregnancy: A Systematic Review and Meta-analysis. Obstet. Gynecol. 2020, 135, 12–23. [Google Scholar] [CrossRef]
- Sebire, N.J.; Fisher, R.A.; Rees, H.C. Histopathological diagnosis of partial and complete hydatidiform mole in the first trimester of pregnancy. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2003, 6, 69–77. [Google Scholar] [CrossRef]
- Ronnett, B.M. Hydatidiform Moles: Ancillary Techniques to Refine Diagnosis. Arch. Pathol. Lab. Med. 2018, 142, 1485–1502. [Google Scholar] [CrossRef]
- Khawajkie, Y.; Mechtouf, N.; Nguyen, N.M.P.; Rahimi, K.; Breguet, M.; Arseneau, J.; Ronnett, B.M.; Hoffner, L.; Lazure, F.; Arnaud, M.; et al. Comprehensive analysis of 204 sporadic hydatidiform moles: Revisiting risk factors and their correlations with the molar genotypes. Mod. Pathol. 2020, 33, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.M.; Fitzgerald, B.; McCarthy, T.V.; Coulter, J.; O’Donoghue, K. Advances in the diagnosis and early management of gestational trophoblastic disease. BMJ Med. 2022, 1, e000321. [Google Scholar] [CrossRef] [PubMed]
- Erol, O.; Süren, D.; Tutuş, B.; Toptaş, T.; Gökay, A.A.; Derbent, A.U.; Özel, M.K.; Sezer, C. Immunohistochemical Analysis of E-Cadherin, p53 and Inhibin-α Expression in Hydatidiform Mole and Hydropic Abortion. Pathol. Oncol. Res. POR 2016, 22, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-R.; Cheng, W.-W.; Wang, Y.-X.; Cai, M.; Wu, W.-B.; Zhang, H.-J. Identification of microRNA signature in the progression of gestational trophoblastic disease. Cell Death Dis. 2018, 9, 94. [Google Scholar] [CrossRef]
- Zheng, X.-Z.; Qin, X.-Y.; Chen, S.-W.; Wang, P.; Zhan, Y.; Zhong, P.-P.; Buza, N.; Jin, Y.-L.; Wu, B.-Q.; Hui, P. Heterozygous/dispermic complete mole confers a significantly higher risk for post-molar gestational trophoblastic disease. Mod. Pathol. 2020, 33, 1979–1988. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, Z.; Jia, C.; Li, J.; Yin, L.; Jiang, S. The relationship between expression of c-ras, c-erbB-2, nm23, and p53 gene products and development of trophoblastic tumor and their predictive significance for the malignant transformation of complete hydatidiform mole. Gynecol. Oncol. 2002, 85, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Yazaki-Sun, S.; Daher, S.; Ishigai, M.M.D.S.; Alves, M.T.S.; Mantovani, T.M.; Mattar, R. Correlation of c-erbB-2 oncogene and p53 tumor suppressor gene with malignant transformation of hydatidiform mole. J. Obstet. Gynaecol. Res. 2006, 32, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Shibata, S.; Oike, A.; Kobayashi, N.; Hamada, H.; Okae, H.; Arima, T. Genomic imprinting in human placentation. Reprod. Med. Biol. 2022, 21, e12490. [Google Scholar] [CrossRef] [PubMed]
- Pasdar, F.A.; Khooei, A.; Fazel, A.; Rastin, M.; Tabasi, N.; Peirouvi, T.; Mahmoudi, M. DNA flow cytometric analysis in variable types of hydropic placentas. Iran. J. Reprod. Med. 2015, 13, 269–274. [Google Scholar]
- Ndukwe, C.O.; Ukah, C.O. Epidemiological Aspects and Diagnostic Accuracy of Morphological Diagnosis of Hydatidiform Mole Using p57kip2 Immunostain in Nnewi, South-East Nigeria—A Multicenter Study. J. Nat. Sci. Med. 2021, 4, 281–287. [Google Scholar]
- Khashaba, M.; Arafa, M.; Elsalkh, E.; Hemida, R.; Kandil, W. Morphological Features and Immunohistochemical Expression of p57Kip2 in Early Molar Pregnancies and Their Relations to the Progression to Persistent Trophoblastic Disease. J. Pathol. Transl. Med. 2017, 51, 381–387. [Google Scholar] [CrossRef][Green Version]
- Lelic, M.; Fatusic, Z.; Iljazovic, E.; Ramic, S.; Markovic, S.; Alicelebic, S. Challenges in the Routine Praxis Diagnosis of Hydatidiform Mole: A Tertiary Health Center Experience. Med. Arch. 2017, 71, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Sasaki, Y.; Kunimura, T.; Sekizawa, A.; Kojima, Y.; Iino, K. Clinical Usefulness of Immunohistochemical Staining of p57 kip2 for the Differential Diagnosis of Complete Mole. BioMed Res. Int. 2015, 2015, 905648. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Adams, E.; Huang, J.; Ronnett, B.M. Refined diagnosis of hydatidiform moles with p57 immunohistochemistry and molecular genotyping: Updated analysis of a prospective series of 2217 cases. Mod. Pathol. 2021, 34, 961–982. [Google Scholar] [CrossRef]
- Chen, K.H.; Hsu, S.C.; Chen, H.Y.; Ng, K.F.; Chen, T.C. Utility of fluorescence in situ hybridization for ploidy and p57 immunostaining in discriminating hydatidiform moles. Biochem. Biophys. Res. Commun. 2014, 446, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Adams, E.; Zou, Y.S.; Morsberger, L.; Scanga, L.R.; Gao, F.F.; Barker, N.; Vang, R.; Ronnett, B.M. Twin/Multiple Gestations with a Hydatidiform Mole: Clinicopathologic Analysis of 21 Cases with Emphasis on Molecular Genotyping and Parental Contribution. Am. J. Surg. Pathol. 2022, 46, 1180–1195. [Google Scholar] [CrossRef] [PubMed]
- Zainal, N.; Kampan, N.C.; Rose, I.M.; Ghazali, R.; Shafiee, M.N.; Yussoff, N.H.; Tamil, A.; Jamil, M.A.; Hussin, N.H. Complementary role of p57kip2 immunostaining in diagnosing hydatidiform mole subtypes. Horm. Mol. Biol. Clin. Investig. 2021, 42, 311–316. [Google Scholar] [CrossRef]
- Takahashi, S.; Okae, H.; Kobayashi, N.; Kitamura, A.; Kumada, K.; Yaegashi, N.; Arima, T. Loss of p57KIP2 expression confers resistance to contact inhibition in human androgenetic trophoblast stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 26606–26613. [Google Scholar] [CrossRef]
- Zheng, X.-Z.; Hui, P.; Chang, B.; Gao, Z.-B.; Li, Y.; Wu, B.-Q.; Zhang, B. STR DNA genotyping of hydatidiform moles in South China. Int. J. Clin. Exp. Pathol. 2014, 7, 4704–4719. [Google Scholar]
- Banet, N.; DeScipio, C.; Murphy, K.M.; Beierl, K.; Adams, E.; Vang, R.; Ronnett, B.M. Characteristics of hydatidiform moles: Analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Mod. Pathol. 2014, 27, 238–254. [Google Scholar] [CrossRef]
- King, J.R.; Wilson, M.L.; Hetey, S.; Kiraly, P.; Matsuo, K.; Castaneda, A.V.; Toth, E.; Krenacs, T.; Hupuczi, P.; Mhawech-Fauceglia, P.; et al. Dysregulation of Placental Functions and Immune Pathways in Complete Hydatidiform Moles. Int. J. Mol. Sci. 2019, 20, 4999. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.F.; Chan, W.Y. The de novo DNA methyltransferase DNMT3A in development and cancer. Epigenetics 2014, 9, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, S.; Wen, S.; Wan, Y.-W.; Peng, H.-H.; Otta, S.; Liu, Z.; Iacovino, M.; Mahen, E.M.; Kyba, M.; Sadikovic, B.; et al. NLRP7 affects trophoblast lineage differentiation, binds to overexpressed YY1 and alters CpG methylation. Hum. Mol. Genet. 2014, 23, 706–716. [Google Scholar] [CrossRef]
- Sanchez-Delgado, M.; Martin-Trujillo, A.; Tayama, C.; Vidal, E.; Esteller, M.; Iglesias-Platas, I.; Deo, N.; Barney, O.; Maclean, K.; Hata, K.; et al. Absence of Maternal Methylation in Biparental Hydatidiform Moles from Women with NLRP7 Maternal-Effect Mutations Reveals Widespread Placenta-Specific Imprinting. PLoS Genet. 2015, 11, e1005644. [Google Scholar] [CrossRef]
- Bolze, P.-A.; Patrier, S.; Cheynet, V.; Oriol, G.; Massardier, J.; Hajri, T.; Guillotte, M.; Bossus, M.; Sanlaville, D.; Golfier, F.; et al. Expression patterns of ERVWE1/Syncytin-1 and other placentally expressed human endogenous retroviruses along the malignant transformation process of hydatidiform moles. Placenta 2016, 39, 116–124. [Google Scholar] [CrossRef]
- Langbein, M.; Strick, R.; Strissel, P.L.; Vogt, N.; Parsch, H.; Beckmann, M.W.; Schild, R.L. Impaired cytotrophoblast cell-cell fusion is associated with reduced Syncytin and increased apoptosis in patients with placental dysfunction. Mol. Reprod. Dev. 2008, 75, 175–183. [Google Scholar] [CrossRef]
- Lertkhachonsuk, R.; Paiwattananupant, K.; Tantbirojn, P.; Rattanatanyong, P.; Mutirangura, A. LINE-1 Methylation Patterns as a Predictor of Postmolar Gestational Trophoblastic Neoplasia. BioMed Res. Int. 2015, 421747, 2015. [Google Scholar] [CrossRef] [PubMed]
- Rahat, B.; Thakur, S.; Bagga, R.; Kaur, J. Epigenetic regulation of STAT5A and its role as fetal DNA epigenetic marker during placental development and dysfunction. Placenta 2016, 44, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Triratanachat, S.; Nakaporntham, P.; Tantbirojn, P.; Shuangshoti, S.; Lertkhachonsuk, R. Role of P57KIP2 Immunohistochemical Expression in Histological Diagnosis of Hydatidiform Moles. Asian Pac. J. Cancer Prev. 2016, 17, 2061–2066. [Google Scholar] [CrossRef][Green Version]
- Samadder, A.; Kar, R. Utility of p57 immunohistochemistry in differentiating between complete mole, partial mole & non-molar or hydropic abortus. Indian J. Med. Res. 2017, 145, 133–137. [Google Scholar] [CrossRef]
- McConnell, T.G.; Murphy, K.M.; Hafez, M.; Vang, R.; Ronnett, B.M. Diagnosis and Subclassification of Hydatidiform Moles Using p57 Immunohistochemistry and Molecular Genotyping: Validation and Prospective Analysis in Routine and Consultation Practice Settings with Development of an Algorithmic Approach. Am. J. Surg. Pathol. 2009, 33, 805–817. [Google Scholar] [CrossRef] [PubMed]
- LeGallo, R.D.; Stelow, E.B.; Ramirez, N.C.; Atkins, K.A. Diagnosis of hydatidiform moles using p57 immunohistochemistry and HER2 fluorescent in situ hybridization. Am. J. Clin. Pathol. 2008, 129, 749–755. [Google Scholar] [CrossRef]
- Diwa, M.H.; Kim, M.A.; Avila, J.M.C.; Pedroza, D.G.; Encinas-Latoy, M.A.M. Utility of p57KIP2 and Her-2 Fluorescence in Situ Hybridization in Differentiating Partial from Complete Hydatidiform Mole. Acta Med. Philipp. 2016, 50, 318–325. [Google Scholar] [CrossRef]
- Frost, J.M.; Moore, G.E. The importance of imprinting in the human placenta. PLoS Genet. 2010, 6, e1001015. [Google Scholar] [CrossRef]
- Tycko, B.; Morison, I.M. Physiological functions of imprinted genes. J. Cell. Physiol. 2002, 192, 245–258. [Google Scholar] [CrossRef]
- Reddy, R.; Akoury, E.; Nguyen, N.M.P.; A Abdul-Rahman, O.; Dery, C.; Gupta, N.; Daley, W.P.; Ao, A.; Landolsi, H.; Fisher, R.A.; et al. Report of four new patients with protein-truncating mutations in C6orf221/KHDC3L and colocalization with NLRP7. Eur. J. Hum. Genet. 2013, 21, 957–964. [Google Scholar] [CrossRef]
- Ji, M.; Shi, X.; Xiang, Y.; Cui, Q.; Zhao, J. NLRP7 and KHDC3L variants in Chinese patients with recurrent hydatidiform moles. Jpn. J. Clin. Oncol. 2019, 49, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.M.P.; Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Curr. Obs. Gynecol. Rep. 2014, 3, 55. [Google Scholar] [CrossRef]
- Nguyen, N.M.P.; Khawajkie, Y.; Mechtouf, N.; Rezaei, M.; Breguet, M.; Kurvinen, E.; Jagadeesh, S.; Solmaz, A.E.; Aguinaga, M.; Hemida, R.; et al. The genetics of recurrent hydatidiform moles: New insights and lessons from a comprehensive analysis of 113 patients. Mod. Pathol. 2018, 31, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Fallahi, J.; Anvar, Z.; Razban, V.; Momtahan, M.; Namavar-Jahromi, B.; Fardaei, M. Founder Effect of KHDC3L, p.M1V Mutation, on Iranian Patients with Recurrent Hydatidiform Moles. Iran. J. Med. Sci. 2020, 45, 118. [Google Scholar] [CrossRef]
- Fallahi, J.; Alashti, S.K.; Aliabadi, B.E.; Mohammadi, S.; Fardaei, M. Recurrent pregnancy loss in the female with a heterozygous mutation in KHDC3L gene. Gene Rep. 2020, 20, 100721. [Google Scholar] [CrossRef]
- Rezaei, M.; Nguyen, N.M.P.; Foroughinia, L.; Dash, P.; Ahmadpour, F.; Verma, I.C.; Slim, R.; Fardaei, M. Two novel mutations in the KHDC3L gene in Asian patients with recurrent hydatidiform mole. Hum. Genome Var. 2016, 3, 16027. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, T.A.; Abdel-Hamid, M.S.; Shaker, M.M. Two Novel Variants in NLRP7 Gene in an Egyptian Female Patient with Consecutive Molar Pregnancies Complicated by Choriocarcinoma. Int. J. Infertil. Fetal Med. 2019, 10, 54–57. [Google Scholar] [CrossRef]
- Fallahi, J.; Razban, V.; Momtahan, M.; Akbarzadeh-Jahromi, M.; Namavar-Jahromi, B.; Anvar, Z. A novel mutation in NLRP7 related to recurrent hydatidiform mole and reproductive failure. Int. J. Fertil. Steril. 2019, 13, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Sethi, P.; Jena, S.K.; Mitra, S. Familial recurrent molar pregnancy: Positive for KHDC3L gene mutation. BMJ Case Rep. 2023, 16, e254435. [Google Scholar] [CrossRef] [PubMed]
- Messaed, C.; Akoury, E.; Djuric, U.; Zeng, J.; Saleh, M.; Gilbert, L.; Seoud, M.; Qureshi, S.; Slim, R. NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J. Biol. Chem. 2011, 286, 43313–43323. [Google Scholar] [CrossRef] [PubMed]
- Sills, E.S.; Obregon-Tito, A.J.; Gao, H.; McWilliams, T.K.; Gordon, A.T.; Adams, C.A.; Slim, R. Pathogenic variant in NLRP7 (19q13.42) associated with recurrent gestational trophoblastic disease: Data from early embryo development observed during in vitro fertilization. Clin. Exp. Reprod. Med. 2017, 44, 40–46. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alici-Garipcan, A.; Özçimen, B.; Süder, I.; Ülker, V.; Önder, T.T.; Özören, N. NLRP7 plays a functional role in regulating BMP4 signaling during differentiation of patient-derived trophoblasts. Cell Death Dis. 2020, 11, 658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, X.; Yu, X.; Huang, B.; Jiang, T.; Zhang, X.; Yang, H.; Qian, J. Abnormal processing of IL-1β in NLRP7-mutated monocytes in hydatidiform mole patients. Clin. Exp. Immunol. 2020, 202, 72–79. [Google Scholar] [CrossRef]
- Reddy, R.; Nguyen, N.M.P.; Sarrabay, G.; Rezaei, M.; Rivas, M.C.G.; Kavasoglu, A.; Berkil, H.; Elshafey, A.; Nunez, K.P.; Dreyfus, H.; et al. The genomic architecture of NLRP7 is Alu rich and predisposes to disease-associated large deletions. Eur. J. Hum. Genet. 2016, 24, 1445–1452. [Google Scholar] [CrossRef][Green Version]
- Rezaei, M.; Suresh, B.; Bereke, E.; Hadipour, Z.; Aguinaga, M.; Qian, J.H.; Bagga, R.; Fardaei, M.; Hemida, R.; Jagadeesh, S.; et al. Novel pathogenic variants in NLRP7, NLRP5, and PADI6 in patients with recurrent hydatidiform moles and reproductive failure. Clin. Genet. 2021, 99, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Buza, N.; McGregor, S.M.; Barroilhet, L.; Zheng, X.; Hui, P. Paternal uniparental isodisomy of tyrosine hydroxylase locus at chromosome 11p15.4: Spectrum of phenotypical presentations simulating hydatidiform moles. Mod. Pathol. 2019, 32, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lu, B.; Lu, W.; Li, S.; Li, X.; Wang, X.; Wan, X.; Chen, Y.; Feng, S.; Jia, Y.; et al. Whole-exome sequencing reveals genetic variants in ERC1 and KCNG4 associated with complete hydatidiform mole in Chinese Han women. Oncotarget 2017, 8, 75264–75271. Available online: https://www.oncotarget.com/article/20769/text/ (accessed on 1 June 2024). [CrossRef] [PubMed][Green Version]
- Hemida, R.; van Doorn, H.; Fisher, R. A Novel Genetic Mutation in a Patient with Recurrent Biparental Complete Hydatidiform Mole: A Brief Report. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2016, 26, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Maehara, K.; Kaneki, E.; Matsuoka, K.; Sugahara, N.; Miyata, T.; Kamura, H.; Yamaguchi, Y.; Kono, A.; Nakabayashi, K.; et al. Novel Nonsense Mutation in the NLRP7 Gene Associated with Recurrent Hydatidiform Mole. Gynecol. Obs. Investig. 2016, 81, 353–358. [Google Scholar] [CrossRef]
- Nguyen, N.M.P.; Ge, Z.-J.; Reddy, R.; Fahiminiya, S.; Sauthier, P.; Bagga, R.; Sahin, F.I.; Mahadevan, S.; Osmond, M.; Breguet, M.; et al. Causative Mutations and Mechanism of Androgenetic Hydatidiform Moles. Am. J. Hum. Genet. 2018, 103, 740–751. [Google Scholar] [CrossRef]
- Al-Jabri, M.; Al-Badi, S.; Al-Kindi, H.; Arafa, M. Immunohistochemical expression of BCL-2 in hydatidiform moles: A tissue microarray study. Pathol. J. Ital. Soc. Anat. Pathol. Diagn. Cytopathol. 2023, 115, 148–154. [Google Scholar] [CrossRef]
- Nguyen, N.M.P.; Zhang, L.; Reddy, R.; Déry, C.; Arseneau, J.; Cheung, A.; Surti, U.; Hoffner, L.; Seoud, M.; Zaatari, G.; et al. Comprehensive genotype-phenotype correlations between NLRP7 mutations and the balance between embryonic tissue differentiation and trophoblastic proliferation. J. Med. Genet. 2014, 51, 623–634. [Google Scholar] [CrossRef]
- Dube, R.; Kar, S.S.; Jhancy, M.; George, B.T. Molecular Basis of Müllerian Agenesis Causing Congenital Uterine Factor Infertility—A Systematic Review. Int. J. Mol. Sci. 2024, 25, 120. [Google Scholar] [CrossRef]
- Khooei, A.; Pasdar, F.A.; Fazel, A.; Mahmoudi, M.; Nikravesh, M.R.; Shahbazian, S.D. View of Expression of Pro-Apoptotic Bax and Anti-Apoptotic Bcl-2 Proteins in Hydatidiform Moles and Placentas with Hydropic Changes. Acta Medica Iran. 2019, 57, 27–32. [Google Scholar] [CrossRef]
- Missaoui, N.; Landolsi, H.; Mestiri, S.; Essakly, A.; Abdessayed, N.; Hmissa, S.; Mokni, M.; Yacoubi, M.T. Immunohistochemical analysis of c-erbB-2, Bcl-2, p53, p21WAF1/Cip1, p63 and Ki-67 expression in hydatidiform moles. Pathol. Res. Pract. 2019, 215, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Erol, O.; Suren, D.; Tutus, B.; Yararbas, K.; Sayiner, A.; Ozel, M.K.; Derbent, A.U.; Sezer, C. Comparison of p57, c-erbB-2, CD117, and Bcl-2 expression in the differential diagnosis of hydatidiform mole and hydropic abortion. Eur. J. Gynaecol. Oncol. 2016, 37, 522–529. Available online: https://pubmed.ncbi.nlm.nih.gov/29894078/ (accessed on 31 May 2024). [PubMed]
- Wang, J.; Zhao, M.; Xiao, J.; Wu, M.; Song, Y.; Yin, Y. E-Cadherin, CD44v6, and Insulin-Like Growth Factor-II mRNA-Binding Protein 3 Expressions in Different Stages of Hydatidiform Moles. J. Biochem. Mol. Toxicol. 2016, 30, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Moussa, R.A.; Eesa, A.N.; Abdallah, Z.F.; Abdelmeged, A.; Mahran, A.; Bahaa, H. Diagnostic Utility of Twist1, Ki-67, and E-Cadherin in Diagnosing Molar Gestations and Hydropic Abortions. Am. J. Clin. Pathol. 2018, 149, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Cicek, O.S.Y.; Hekimoglu, E.R.; Turgal, M.; Atilla, P.; Cakar, A.N.; Usubutun, A.; Beksac, M.S. Differential expression of leukemia inhibitory factor and insulin like growth factor-1 between normal pregnancies, partial hydatidiform moles and complete hydatidiform moles. Placenta 2018, 69, 64–70. [Google Scholar] [CrossRef]
- Deka FF, H.A.; Abd Ali Al Saeng, Z.H.; Almukhtar, K. Role of the Immunohistochemical Marker (Ki67) in Diagnosis and Classification of Hydatidiform Mole. IIUM Med. J. Malays. 2019, 18, 136–142. [Google Scholar] [CrossRef]
- Guo, Z.; Sui, L.; Qi, J.; Sun, Q.; Xu, Y.; Zou, N.; Xie, Y.; Kong, Y. miR-196b inhibits cell migration and invasion through targeting MAP3K1 in hydatidiform mole. Biomed. Pharmacother. 2019, 113, 108760. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhao, J.R.; Xu, Y.Y.; Wu, W.B.; Zhang, H.J. miR-21 Is Overexpressed in Hydatidiform Mole Tissues and Promotes Proliferation, Migration, and Invasion in Choriocarcinoma Cells. Int. J. Gynecol. Cancer 2017, 27, 364–374. [Google Scholar] [CrossRef]
- Kheradmand, P.; Goudarzi, M.; Tavakoli, M. Analysis of p53 expression in partial hydatidiform mole and hydropic abortion. Front. Biol. 2017, 12, 357–360. [Google Scholar] [CrossRef]
- Khooei, A.; Pasdar, F.A.; Fazel, A.; Mahmoudi, M.; Nikravesh, M.R.; Shahbazian, S.D. P53 expression in various types of hydropic placentas (through ploidy analysis as a complementary tool in diagnosis of samples). Casp. J. Intern. Med. 2019, 10, 205. [Google Scholar] [CrossRef]
- Kubelka-Sabit, K.; Prodanova, I.; Jasar, D.; Bozinovski, G.; Filipovski, V.; Drakulevski, S.; Plaseska-Karanfilska, D. Molecular and immunohistochemical characteristics of complete hydatidiform moles. Balk. J. Med. Genet. 2017, 20, 27–34. [Google Scholar] [CrossRef][Green Version]
- Masood, S.; Kehar, S.I.; Shawana, S.; Aamir, I. Differential expression of p63 in hydropic and molar gestations. J. Coll. Physicians Surg. Pak. 2015, 25, 198–202. [Google Scholar] [PubMed]
- Jahanbin, B.; Sarmadi, S.; Ghasemi, D.; Nili, F.; Moradi, J.-A.; Ghasemi, S. Pathogenic role of Twist-1 protein in hydatidiform molar pregnancies and investigation of its potential diagnostic utility in complete moles. Diagn. Pathol. 2023, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Parcesepe, P.; Mafficini, A.; Nottegar, A.; Parolini, C.; Veronese, N.; Remo, A.; Manfrin, E. Specific expression patterns of epithelial to mesenchymal transition factors in gestational molar disease. Placenta 2015, 36, 1318–1324. [Google Scholar] [CrossRef]
- Hasanzadeh, M.; Sharifi, N.; Farazestanian, M.; Nazemian, S.S.; Sani, F.M. Immunohistochemistry Study of P53 and C-erbB-2 Expression in Trophoblastic Tissue and Their Predictive Values in Diagnosing Malignant Progression of Simple Molar Pregnancy. Iran. J. Cancer Prev. 2016, 9, e4115. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Wu, Q.; Ruan, G.; Zheng, X.; Song, Y.; Zhun, J.; Wu, L.; Gotlieb, W.H. Expression patterns of maspin and mutant p53 are associated with the development of gestational trophoblastic neoplasia. Oncol. Lett. 2016, 12, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Hadi, F.; Kazemi, N.; Hosseini, M.S.; Ebrahimi, A. Evaluation of TP53 and HER-2/neu Genes Expression Levels in Gestational Trophoblastic Diseases Cases and Determining Their Predictive Value in Diagnosis of Malignancy and Disease Progression. Int. J. Cancer Manag. 2022, 15, 119264. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Mak, V.C.; Lee, L.; Siu, M.K.; Wong, O.G.; Lu, X.; Ngan, H.Y.; Wong, E.S.; Cheung, A.N. Downregulation of ASPP2 in choriocarcinoma contributes to increased migratory potential through Src signaling pathway activation. Carcinogenesis 2013, 34, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- Mak, V.C.Y.; Lee, L.; Siu, M.K.Y.; Wong, O.G.W.; Lu, X.; Ngan, H.Y.S.; Wong, E.S.Y.; Cheung, A.N.Y. Downregulation of ASPP1 in gestational trophoblastic disease: Correlation with hypermethylation, apoptotic activity and clinical outcome. Mod. Pathol. 2011, 24, 522–532. [Google Scholar] [CrossRef]
- Chan, K.-K.; Wong, E.S.-Y.; Wong, I.T.-L.; Cheung, C.L.-Y.; Wong, O.G.-W.; Ngan, H.Y.-S.; Cheung, A.N.-Y. Overexpression of iASPP is required for autophagy in response to oxidative stress in choriocarcinoma. BMC Cancer 2019, 19, 953. [Google Scholar] [CrossRef]
- Chan, K.K.; Wong, E.S.Y.; Wong, O.G.W.; Ngan, H.Y.S.; Cheung, A.N.Y. Identification of nonsynonymous TP53 mutations in hydatidiform moles. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2018, 809, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Mishra, C.; Biswal, P.; Kar, T.; Panda, S.; Naik, S. Differential expression of cyclin E, p63, and Ki-67 in gestational trophoblastic disease and its role in diagnosis and management: A prospective case-control study. Indian J. Pathol. Microbiol. 2019, 62, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Braga, A.; Maestá, I.; Soares, R.R.; Elias, K.M.; Domingues, M.A.C.; Barbisan, L.F.; Berkowitz, R.S. Apoptotic index for prediction of postmolar gestational trophoblastic neoplasia. Am. J. Obstet. Gynecol. 2016, 215, 336.e1–336.e12. [Google Scholar] [CrossRef] [PubMed]
- Toki, T.; Horiuchi, A.; Ichikawa, N.; Mori, A.; Nikaido, T.; Fujii, S. Inverse relationship between apoptosis and Bcl-2 expression in syncytiotrophoblast and fibrin-type fibrinoid in early gestation. Mol. Hum. Reprod. 1999, 5, 246–251. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagib, R.M.; MAZaki, M.; Wageh, A.; Abdelrazik, M. Can Ki67 and Caspase Predict Molar Progression? Fetal Pediatr. Pathol. 2019, 38, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Ronnett, B.M.; Descipio, C.; Murphy, K.M. Hydatidiform moles: Ancillary techniques to refine diagnosis. Int. J. Gynecol. Pathol. 2011, 30, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Wargasetia, T.L.; Shahib, M.N.; Martaadisoebrata, D.; Dhianawaty, D.; Hernowo, B. Characterization of apoptosis and autophagy through Bcl-2 and Beclin-1 immunoexpression in gestational trophoblastic disease. Iran. J. Reprod. Med. 2015, 13, 413. [Google Scholar] [PubMed]
- Lin, L.H.; Maestá, I.; Laurent, J.D.S.; Hasselblatt, K.T.; Horowitz, N.S.; Goldstein, D.P.; Quade, B.J.; Sun, S.Y.; Braga, A.; Fisher, R.A.; et al. Distinct microRNA profiles for complete hydatidiform moles at risk of malignant progression. Am. J. Obstet. Gynecol. 2021, 224, 372.e1–372.e30. [Google Scholar] [CrossRef]
- Fulop, V.; Mok, S.C.; Genest, D.R.; Szigetvari, I.; Cseh, I.; Berkowitz, R.S. c-myc, c-erbB-2, c-fms and bcl-2 oncoproteins. Expression in normal placenta, partial and complete mole, and choriocarcinoma. J. Reprod. Med. 1998, 43, 101–110. [Google Scholar]
- Al-Bozom, I.A. P53 and Bcl-2 oncoprotein expression in placentas with hydropic changes and partial and complete moles. APMIS 2000, 108, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Pinkas-Kramarski, R.; Alroy, I.; Yarden, Y. ErbB receptors and EGF-like ligands: Cell lineage determination and oncogenesis through combinatorial signaling. J. Mammary Gland. Biol. Neoplasia 1997, 2, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Fock, V.; Plessl, K.; Fuchs, R.; Dekan, S.; Milla, S.K.; Haider, S.; Fiala, C.; Knöfler, M.; Pollheimer, J. Trophoblast subtype-specific EGFR/ERBB4 expression correlates with cell cycle progression and hyperplasia in complete hydatidiform moles. Hum. Reprod. 2015, 30, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. KIT (CD117): A review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 205–220. [Google Scholar] [CrossRef]
- Sheikh, E.; Tran, T.; Vranic, S.; Levy, A.; Bonfil, R.D. Role and significance of c-KIT receptor tyrosine kinase in cancer: A review. Bosn. J. Basic Med. Sci. 2022, 22, 683. [Google Scholar] [CrossRef] [PubMed]
- Pekcan, M.K.; Tokmak, A.; Topfedaisi Ozkan, N.; Ozaksit, G.; Kosem, A.; Erel, O.; Meydanli, M. Thiol/Disulfide Homeostasis in Patients with Molar Pregnancies. Fetal Pediatr. Pathol. 2020, 39, 99–106. [Google Scholar] [CrossRef]
- Incebiyik, A.; Vural, M.; Camuzcuoglu, H.; Taskin, A.; Camuzcuoglu, A.; Hilali, N.G.; Aksoy, N. Can circulating M30 and M65 levels be beneficial markers in the diagnosis and management of patients with complete hydatidiform mole? Wien. Klin. Wochenschr. 2016, 128 (Suppl 8), 566–571. [Google Scholar] [CrossRef]
- Agarwal, A.; Gupta, S.; Sharma, R.K. Role of oxidative stress in female reproduction. Reprod. Biol. Endocrinol. 2005, 3, 28–48. [Google Scholar] [CrossRef]
- Martinez, C.; González-Ramírez, J.; Marín, M.E.; Martínez-Coronilla, G.; Meza-Reyna, V.I.; Mora, R.; Díaz-Molina, R. Isthmin 2 is decreased in preeclampsia and highly expressed in choriocarcinoma. Heliyon 2020, 6, e05096. [Google Scholar] [CrossRef]
- Chiang, Y.T.; Seow, K.M.; Chen, K.H. The Pathophysiological, Genetic, and Hormonal Changes in Preeclampsia: A Systematic Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2024, 25, 4532. [Google Scholar] [CrossRef]
- Rayner, A.A.; Berkowitz, R.; Steele, G.; Schur, P.H.; Rodrick, M.L.; Goldstein, D.P.; Harte, P.J.; Wilson, R.E.; Zamcheck, N.; Munroe, A.E. Circulating immune complex levels in patients with gestational trophoblastic neoplasia. J. Natl. Cancer Inst. 1982, 69, 23–26. Available online: https://pubmed.ncbi.nlm.nih.gov/6285061/ (accessed on 1 June 2024). [PubMed]
- Piani, F.; Tossetta, G.; Fantone, S.; Agostinis, C.; Di Simone, N.; Mandalà, M.; Bulla, R.; Marzioni, D.; Borghi, C. First Trimester CD93 as a Novel Marker of Preeclampsia and Its Complications: A Pilot Study. High Blood Press. Cardiovasc. Prev. Off. J. Ital. Soc. Hypertens. 2023, 30, 591–594. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).