
Mitogenomes of Three Satyrid Butterfly Species (Nymphalidae: Lepidoptera) and Reconstructed Phylogeny of Satyrinae


Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimens and DNA Extraction
2.2. Mitogenome Sequencing, Assembly, and Annotation
2.3. Sequence Analyses
2.4. Divergence Time Estimation
2.5. Phylogenetic Analysis
3. Results and Discussion
3.1. General Mitogenomic Features
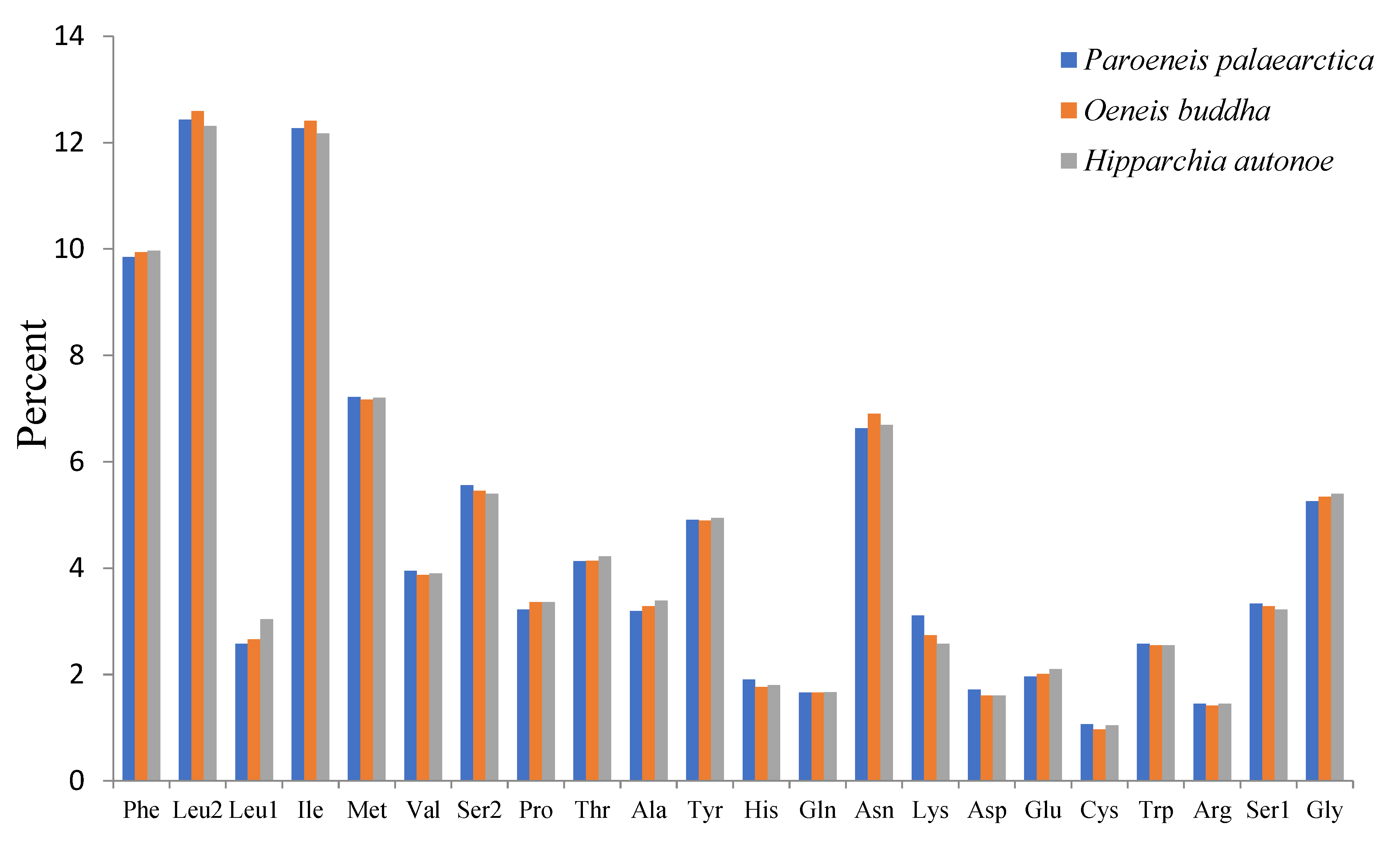
3.2. Protein Coding Genes and Codon Usage
3.3. The rRNAs and tRNAs
3.4. Intergenic and Overlapping Spacer Regions
3.5. A + t-Rich Region
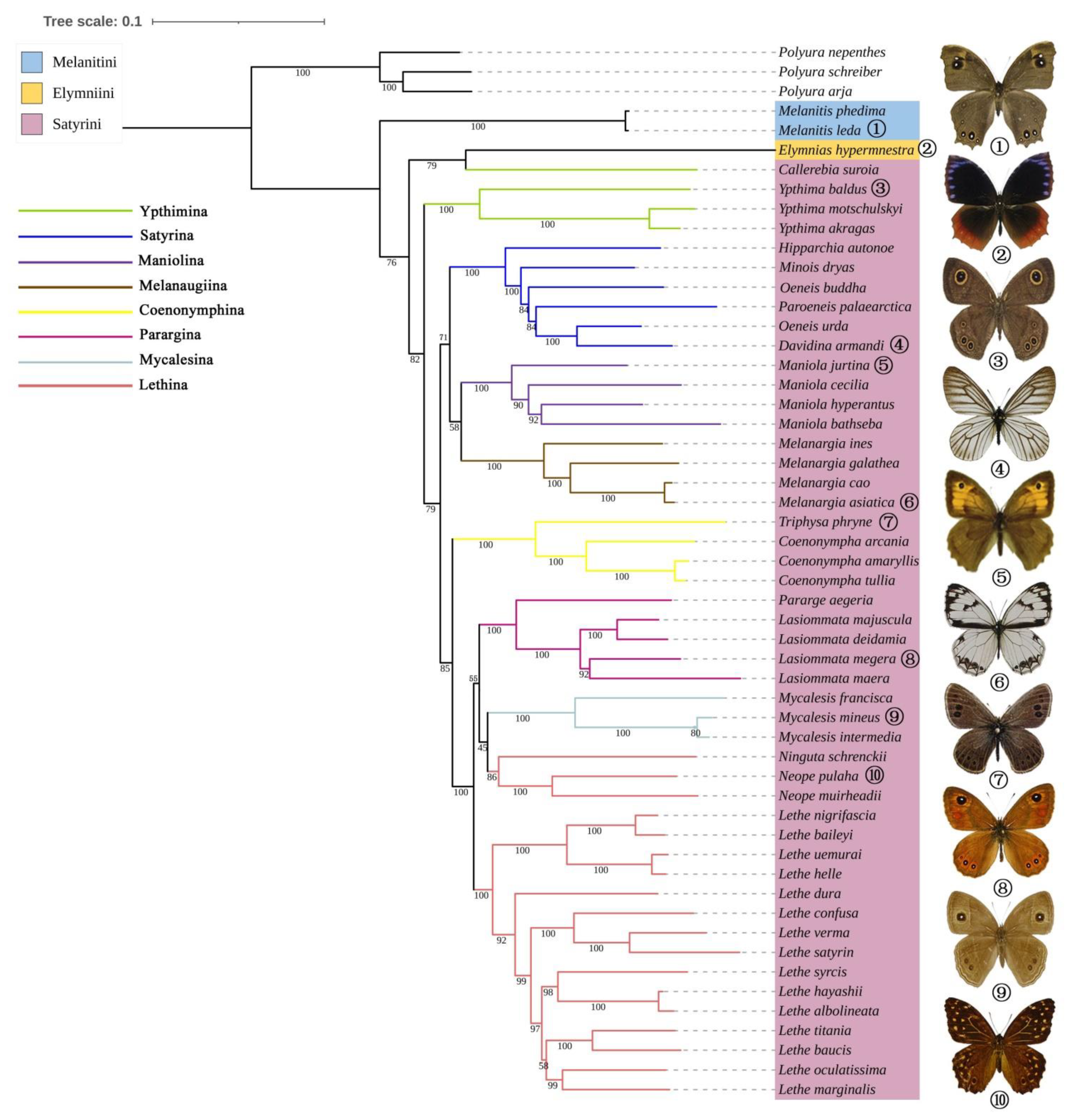
3.6. Phylogenetic Analyses
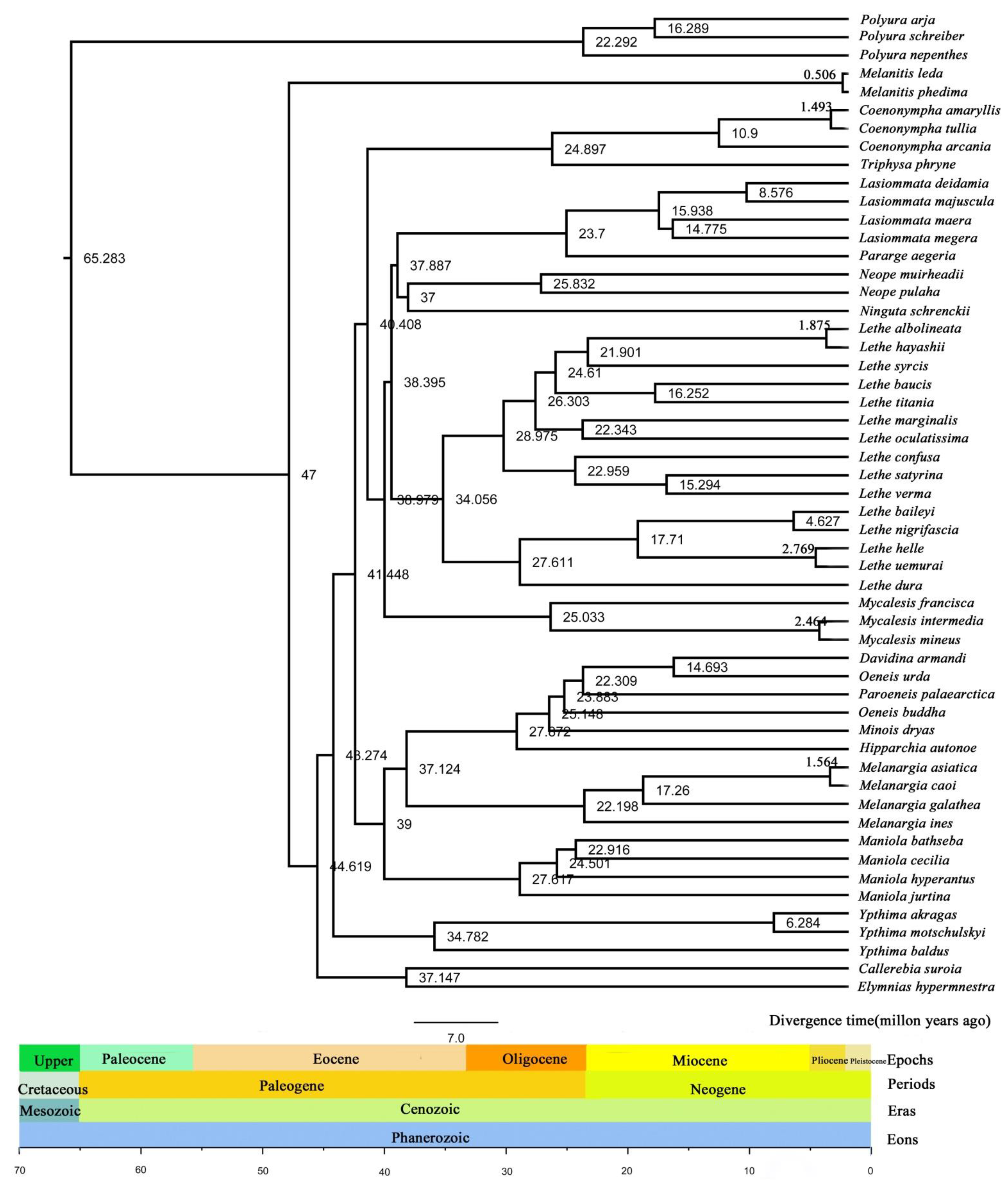
3.7. Divergence Time Estimation of Satyrinae Species
- (1)
- When the phylogenetic tree is reconstructed, the choice of sequence data will inevitably have a certain impact on the research results, because of the difference in the evolution rate of different sequences. This study used mitochondrial genome protein-coding genes for phylogenetic tree reconstruction and molecular clock analysis, while previous studies all used partial mitochondrial genome sequence fragments or mitochondrial partial genes combined with one or two nuclear genes to reconstruct the phylogenetic tree. The molecular clock analysis results are slightly different.
- (2)
- The results of the study are affected by the different embedding locations of the fossil correction points and the different setting of the time during molecular clock calculation.
- (3)
- The value given by the molecular clock calculation is the average value of a confidence interval, that is, the divergence time of each clade in the research results is not a given value but a time range, and the data results given only provide convenience for intuitive comparison.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ackery, P.R.; de Jong, R.; Vane-Wright, R.I. The Butterflies: Hedyloidea, Hesperioidea and Papilionoidea. In Handbook of Zoology; Vol. IV Arthropoda: Insecta. Lepidoptera, Moths and Butterflies, vol. 1: Evolution, Systematics and Biogeography; Kristensen, N.P., Ed.; Walter de Gruyter: Berlin, Germany, 1999; Volume 4, pp. 263–300. [Google Scholar]
- Peña, C.; Wahlberg, N. Prehistorical climate change increased diversification of a group of butterflies. Biol. Lett. 2008, 4, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, C.; Wahlberg, N.; Weingartner, E.; Kodandaramaiah, U.; Brower, A.V.Z. Higher level phylogeny of Satyrinae butterflies (Lepidoptera: Nymphalidae) based on DNA sequence data. Mol. Phylogenetics Evol. 2006, 40, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, N.; Leneveu, J.; Kodandaramaiah, U.; Peña, C.; Nylin, S.; Freitas, A.V.; Brower, A.V. Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. Proc. Biol. Sci. 2009, 276, 4295–4302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, C.; Nylin, S.; Wahlberg, N. The radiation of Satyrini butterflies (Nymphalidae: Satyrinae): A challenge for phylogenetic methods. Zool. J. Linn. Soc. 2011, 161, 64–87. [Google Scholar] [CrossRef] [Green Version]
- Marin, M.A.; Peña, C.; Freitas, A.V.; Wahlberg, N.; Uribe, S.I. From the Phylogeny of the Satyrinae Butterflies to the Systematics of Euptychiina (Lepidoptera: Nymphalidae): History, Progress and Prospects. Neotrop. Entomol. 2011, 40, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.; Wan, X.; Min, J.K.; Kim, I. Genetic relationships between Oeneis urda and O. mongolica (Nymphalidae: Lepidoptera). Entomol. Res. 2013, 43, 85–100. [Google Scholar] [CrossRef]
- Leech, J.H. Butterflies from China, Japan and Corea; RH Porter: London, UK, 1892; pp. 1–681. [Google Scholar]
- Kuznezov, N. On the systematic position of the genus Davidina Obth. (Lepidoptera, Papilionoidea). In Annuaire du Musée Zoologique de l’Académie des Sciences de l’URSS; Académie des Sciences de l’URSS: Moscow, Russia, 1930; pp. 347–358. [Google Scholar]
- Chou, I. Monographia Rhaopalocerorum Sinensium (Revised Edition); Henan Scientific and Technological Publishing House: Zhengzhou, China, 2000. [Google Scholar]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, M.; Lakra, W.S.; Mohindra, V.; Khare, P.; Barman, A.S.; Singh, A.; Lal, K.K.; Punia, P.; Khan, A.A. Evaluation of cytochrome b mtDNA sequences in genetic diversity studies of Channa marulius (Channidae: Perciformes). Mol. Biol. Rep. 2011, 38, 841–846. [Google Scholar] [CrossRef]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating Molecular Evolution into Phylogenetic Analysis, and a New Compilation of Conserved Polymerase Chain Reaction Primers for Animal Mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545. [Google Scholar] [CrossRef] [Green Version]
- Salvato, P.; Simonato, M.; Battisti, A.; Negrisolo, E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genom. 2008, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect Mitochondrial Genomics: Implications for Evolution and Phylogeny. Annu. Rev. Èntomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, M.J.; Lees, D.C.; Simonsen, T.J. Towards a mitogenomic phylogeny of Lepidoptera. Mol. Phylogenetics Evol. 2014, 79, 169–178. [Google Scholar] [CrossRef]
- Li, J.; Xu, C.; Lei, Y.; Fan, C.; Gao, Y.; Xu, C.; Wang, R. Complete mitochondrial genome of a satyrid butterfly, Lethe albolineata (Lepidoptera: Nymphalidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 4195–4196. [Google Scholar] [CrossRef]
- Zhang, W.; Gan, S.; Zuo, N.; Chen, C.; Wang, Y.; Hao, J. The complete mitochondrial genome of Triphysa phryne (Lepidoptera: Nymphalidae: Satyrinae). Mitochondrial DNA 2016, 27, 474–475. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, W.; Hao, J. The complete mitochondrial genome of Callerebia suroia (Lepidoptera: Nymphalidae: Satyrinae). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2014, 27, 1–3. [Google Scholar] [CrossRef]
- Fan, C.; Xu, C.; Li, J.; Lei, Y.; Gao, Y.; Xu, C.; Wang, R. Complete mitochondrial genome of a satyrid butterfly, Ninguta schrenkii (Lepidoptera: Nymphalidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Hao, J.; Zhang, W.; Su, T.; Xu, X. The complete mitochondrial genome of Melanargia asiatica (Lepidoptera: Nymphalidae: Satyrinae). Mitochondrial DNA 2014, 27, 1–3. [Google Scholar] [CrossRef]
- Shi, Q.-H.; Lin, X.-Q.; Ye, X.; Xing, J.-H.; Dong, G.-W. Characterization of the complete mitochondrial genome of Minois dryas (Lepidoptera: Nymphalidae: Satyrinae) with phylogenetic analysis. Mitochondrial DNA Part B 2019, 4, 1447–1449. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.W.; Lin, L.H.; Lees, D.C.; Hsu, Y.F. Mitogenomic sequences effectively recover relationships within brush-footed butterflies (Lepidoptera: Nymphalidae). BMC Genom. 2014, 15, 468. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.-H.; Sun, X.-Y.; Wang, Y.-L.; Hao, J.-S.; Yang, Q. Morphological Characters Are Compatible with Mitogenomic Data in Resolving the Phylogeny of Nymphalid Butterflies (Lepidoptera: Papilionoidea: Nymphalidae). PLoS ONE 2015, 10, e0124349. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Song, L.; Zhou, L.; Shi, Y.; Song, N.; Zhang, Y. Mitochondrial genomes of four satyrine butterflies and phylogenetic relationships of the family Nymphalidae (Lepidoptera: Papilionoidea). Int. J. Biol. Macromol. 2019, 145, 272–281. [Google Scholar] [CrossRef]
- Chen, L.; Wahlberg, N.; Liao, C.Q.; Wang, C.B.; Ma, F.Z.; Huang, G.H. Fourteen complete mitochondrial genomes of butterflies from the genus Lethe (Lepidoptera, Nymphalidae, Satyrinae) with mitogenome-based phylogenetic analysis. Genomics 2020, 112, 4435–4441. [Google Scholar] [CrossRef]
- Liu, G.; Chang, Z.; Chen, L.; He, J.; Li, X. Genome size variation in butterflies (Insecta, Lepidotera, Papilionoidea): A thorough phylogenetic comparison. Syst. Èntomol. 2020, 45, 571–582. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, C.; Zhai, Q.; Zhang, Y. The complete mitochondrial genome sequence of Coenonympha amaryllis and monophyly of Satyrinae (Lepidoptera: Nymphalidae). Mitochondrial DNA Part B 2020, 5, 1223–1224. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.H.; Zhao, F.; Hao, J.S.; Yang, Q. Complete mitochondrial genome of the Common Evening Brown, Melanitis leda Linnaeus (Lepidoptera: Nymphalidae: Satyrinae). Mitochondrial DNA 2013, 24, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-P.; Lu, J.-J.; Yang, J.; Wang, J.-P.; Cao, T.-W.; Fan, R.-J. Complete mitochondrial genome of Mycalesis intermedia (Lepidoptera: Nymphalidae). Mitochondrial DNA Part B 2020, 5, 703–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, T.; Tan, M.; Meng, G.; Yang, S.; Zhou, X. Multiplex sequencing of pooled mitochondrial genomes—a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res. 2014, 42, e166. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Z.; Wang, S.; Zhong, H.; Wang, N.; Liang, B. A mitogenomic phylogeny of satyrid butterflies and complete mitochondrial genome of Oeneis urda (Lepidoptera: Nymphalidae: Satyrinae). Mitochondrial DNA Part B 2020, 5, 1344–1345. [Google Scholar] [CrossRef]
- Teixeira da Costa, L.F. The complete mitochondrial genome of Parage aegeria (Insecta: Lepidoptera: Papilionidae). Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Hu, H.W.; Zhang, S.L.; Wang, J.W.; Li, R. Mitochondrial DNA Part B Characterization of the complete mitochondrial genome of Ypthima baldus (Lepidoptera: Satyrinae) with phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 1019–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cong, Q.; Shen, J.; Opler, P.A.; Grishin, N.V. Genomics of a complete butterfly continent. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Min-Shan Ko, A.; Zhang, Y.; Yang, M.A.; Hu, Y.; Cao, P.; Feng, X.; Zhang, L.; Wei, F.; Fu, Q. Mitochondrial genome of a 22,000-year-old giant panda from southern China reveals a new panda lineage. Curr. Biol. CB 2018, 28, R693–R694. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Wiemers, M.; Chazot, N.; Wheat, C.W.; Schweiger, O.; Wahlberg, N. A complete time-calibrated multi-gene phylogeny of the European butterflies. ZooKeys 2020, 938, 97–124. [Google Scholar] [CrossRef]
- Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772. [Google Scholar]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, Y. Phylogenetic utility of ribosomal genes for reconstructing the phylogeny of five Chinese satyrine tribes (Lepidoptera, Nymphalidae). ZooKeys 2015, 488, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Lukhtanov, V.A.; Dubatolov, V.V. Phylogenetic position and taxonomic rearrangement of Davidina (Lepidoptera: Nymphalidae), an enigmatic butterfly genus new for Europe and America. Zool. J. Linn. Soc. 2020, 190, 1036–1053. [Google Scholar] [CrossRef]
- Usami, S.I.; Isaka, Y.; Nishio, S.-Y.; Nakatani, T.; Itoh, T. Phylogeny and biogeography of arctic-alpine butterflies of the genus Oeneis (Nymphalidae: Satyrinae). Èntomol. Sci. 2021, 24, 183–195. [Google Scholar] [CrossRef]
- Lukhtanov, V.A.; Eitschberger, U. Catalogue of the genera Oeneis and Davidina. In Butterflies of the World Supplement 4; Bauer, E., Frankenbach, T., Eds.; Goecke & Evers: Keltern, Germany, 2001. [Google Scholar]
- Heikkila, M.; Kaila, L.; Mutanen, M.; Pena, C.; Wahlberg, N. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. Biol. Sci. 2012, 279, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.P.; Robbins, R.K.; Harvey, D.J. Extinction and biogeography in the Caribbean: New evidence from a fossil riodinid butterfly in Dominican amber. Proc. Biol. Sci. 2004, 271, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Q.; Chen, Z.; Guan, D.; Qiao, F.; Xu, S. Phylogeny and divergent time of Butterflies based on COΙ,Cyt b and EF-Ια genes. J. Shaanxi Norm. Univ. Nat. Sci. Ed. 2019, 47, 108–116. [Google Scholar]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Espeland, M.; Breinholt, J.; Willmott, K.R.; Warren, A.D.; Vila, R.; Toussaint, E.; Maunsell, S.C.; Aduse-Poku, K.; Talavera, G.; Eastwood, R. A Comprehensive and Dated Phylogenomic Analysis of Butterflies. Curr. Biol. 2018, 28, 770–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Size | A + T% | AT-Skew | GC-Skew | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hip | Par | Oen | Hip | Par | Oen | Hip | Par | Oen | Hip | Par | Oen | |
| Whole genome | 15,435 | 15,942 | 15,259 | 79.1 | 78.2 | 79.6 | −0.017 | −0.022 | −0.029 | −0.238 | −0.179 | −0.225 |
| Protein coding genes | 11,208 | 11,212 | 11,205 | 76.8 | 78.8 | 781 | −0.156 | −0.157 | −0.157 | −0.007 | 0.026 | −0.002 |
| 1st codon position | 3735 | 3736 | 3734 | 72.4 | 73.2 | 73.1 | −0.007 | 0 | −0.001 | 0.186 | 0.195 | 0.194 |
| 2nd codon position | 3735 | 3736 | 3734 | 70.1 | 70.3 | 70.3 | −0.381 | −0.371 | −0.379 | −0.089 | −0.081 | −0.089 |
| 3rd codon position | 3735 | 3736 | 3734 | 87.8 | 92.8 | 90.7 | −0.098 | −0.121 | −0.111 | −0.245 | −0.167 | −0.293 |
| tRNA genes | 1442 | 1452 | 1450 | 80.6 | 80.6 | 80.3 | 0.015 | 0.017 | 0.013 | 0.164 | 0.163 | 0.172 |
| rRNA genes | 1358 | 1362 | 1362 | 83.5 | 81.3 | 84.0 | 0.095 | 0.064 | 0.078 | 0.339 | 0.265 | 0.355 |
| A + T-rich region | 896 | 1039 | 452 | 94.6 | 61.8 | 91.4 | −0.016 | −0.003 | −0.026 | −0.208 | −0.003 | 0.076 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dan, Z.; Duan, L.; Chen, Z.; Guan, D.; Xu, S. Mitogenomes of Three Satyrid Butterfly Species (Nymphalidae: Lepidoptera) and Reconstructed Phylogeny of Satyrinae. Diversity 2021, 13, 468. https://doi.org/10.3390/d13100468
Dan Z, Duan L, Chen Z, Guan D, Xu S. Mitogenomes of Three Satyrid Butterfly Species (Nymphalidae: Lepidoptera) and Reconstructed Phylogeny of Satyrinae. Diversity. 2021; 13(10):468. https://doi.org/10.3390/d13100468
Chicago/Turabian StyleDan, Zhicuo, Lei Duan, Zhenning Chen, Delong Guan, and Shengquan Xu. 2021. "Mitogenomes of Three Satyrid Butterfly Species (Nymphalidae: Lepidoptera) and Reconstructed Phylogeny of Satyrinae" Diversity 13, no. 10: 468. https://doi.org/10.3390/d13100468
APA StyleDan, Z., Duan, L., Chen, Z., Guan, D., & Xu, S. (2021). Mitogenomes of Three Satyrid Butterfly Species (Nymphalidae: Lepidoptera) and Reconstructed Phylogeny of Satyrinae. Diversity, 13(10), 468. https://doi.org/10.3390/d13100468