
TFNetPropX: A Web-Based Comprehensive Analysis Tool for Exploring Condition-Specific RNA-Seq Data Using Transcription Factor Network Propagation
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
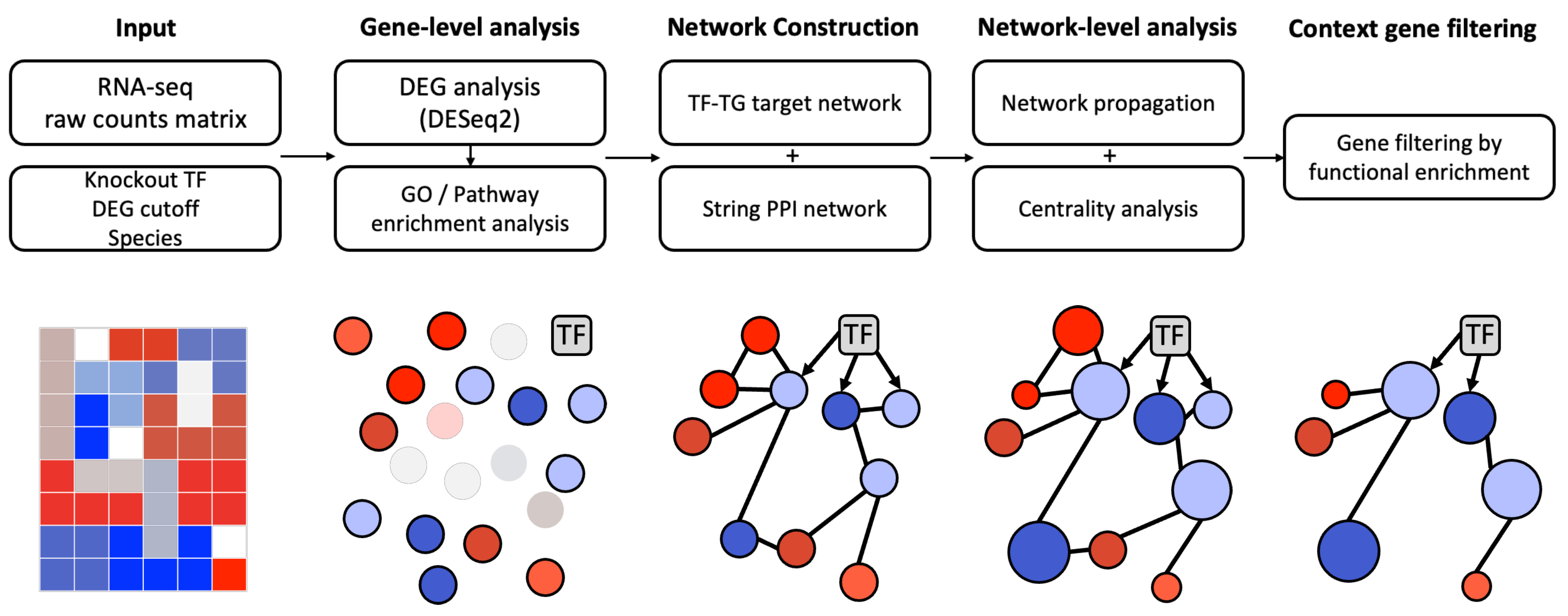
2. Materials and Methods
2.1. Step 1: Input
2.2. Step 2: Gene-Level Analysis
2.3. Step 3: Network Construction
2.4. Step 4: Network Analysis
2.5. Step 5: Context-Specific Gene Filtering
2.6. Step 6: Result Page Generation
3. Results
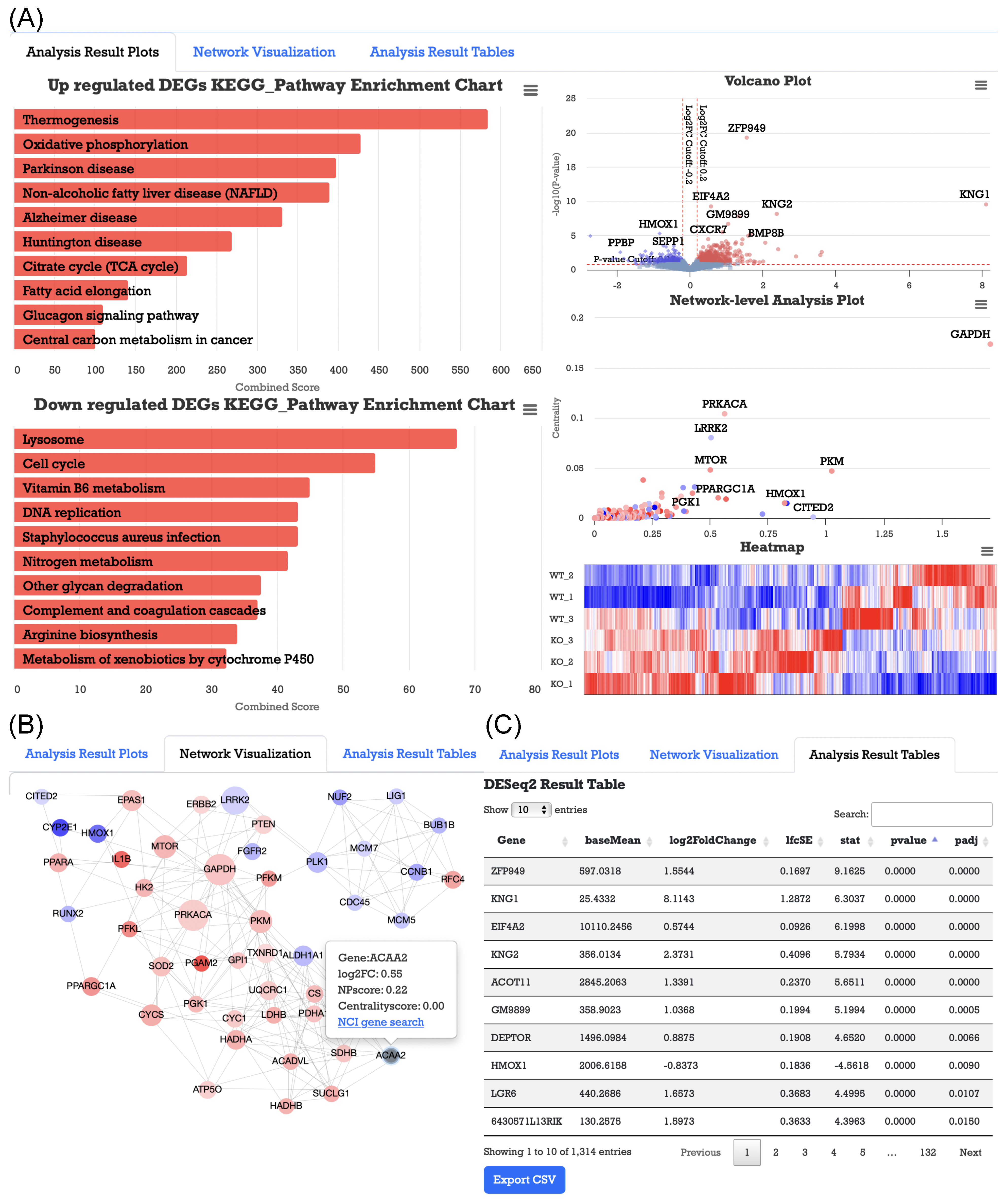
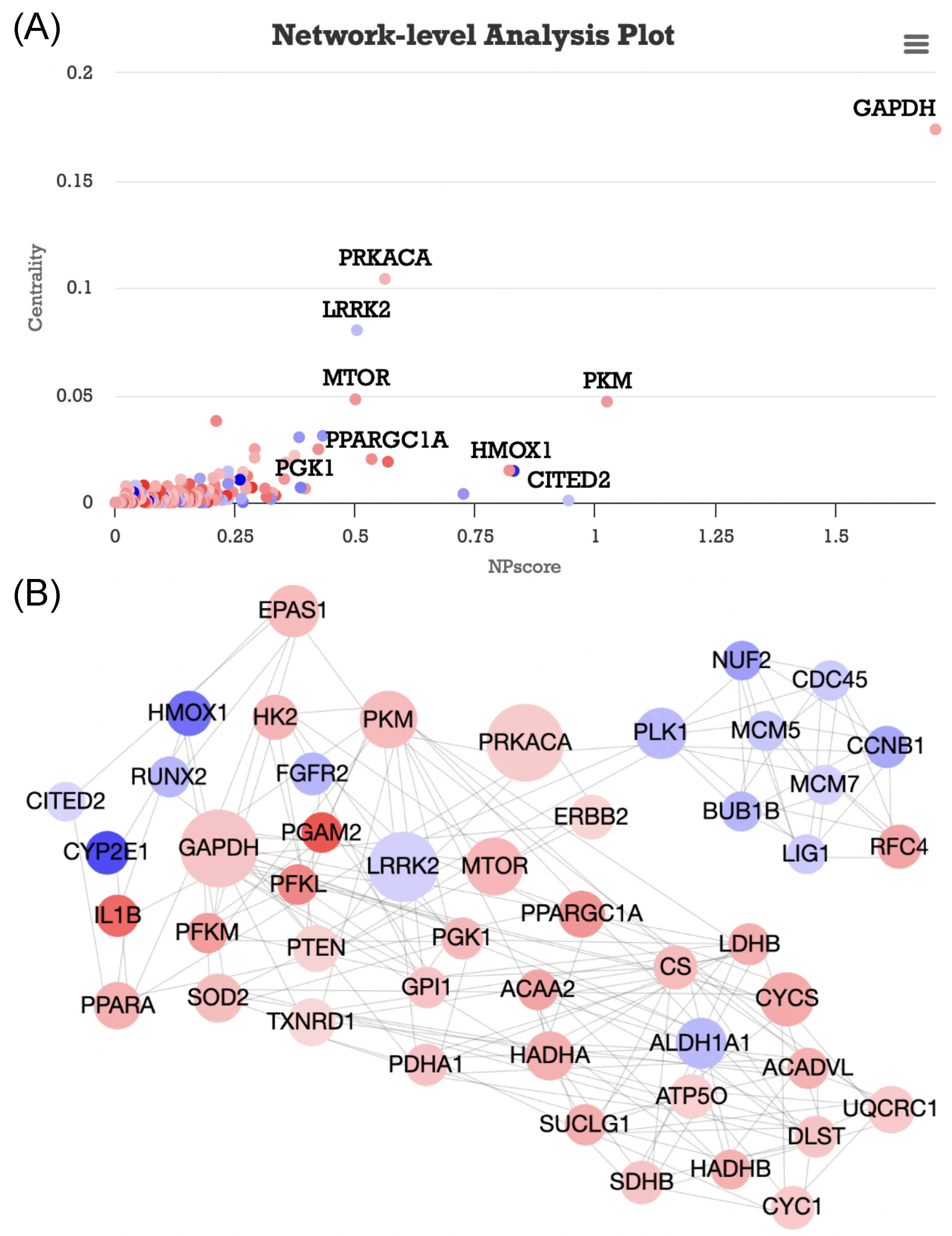
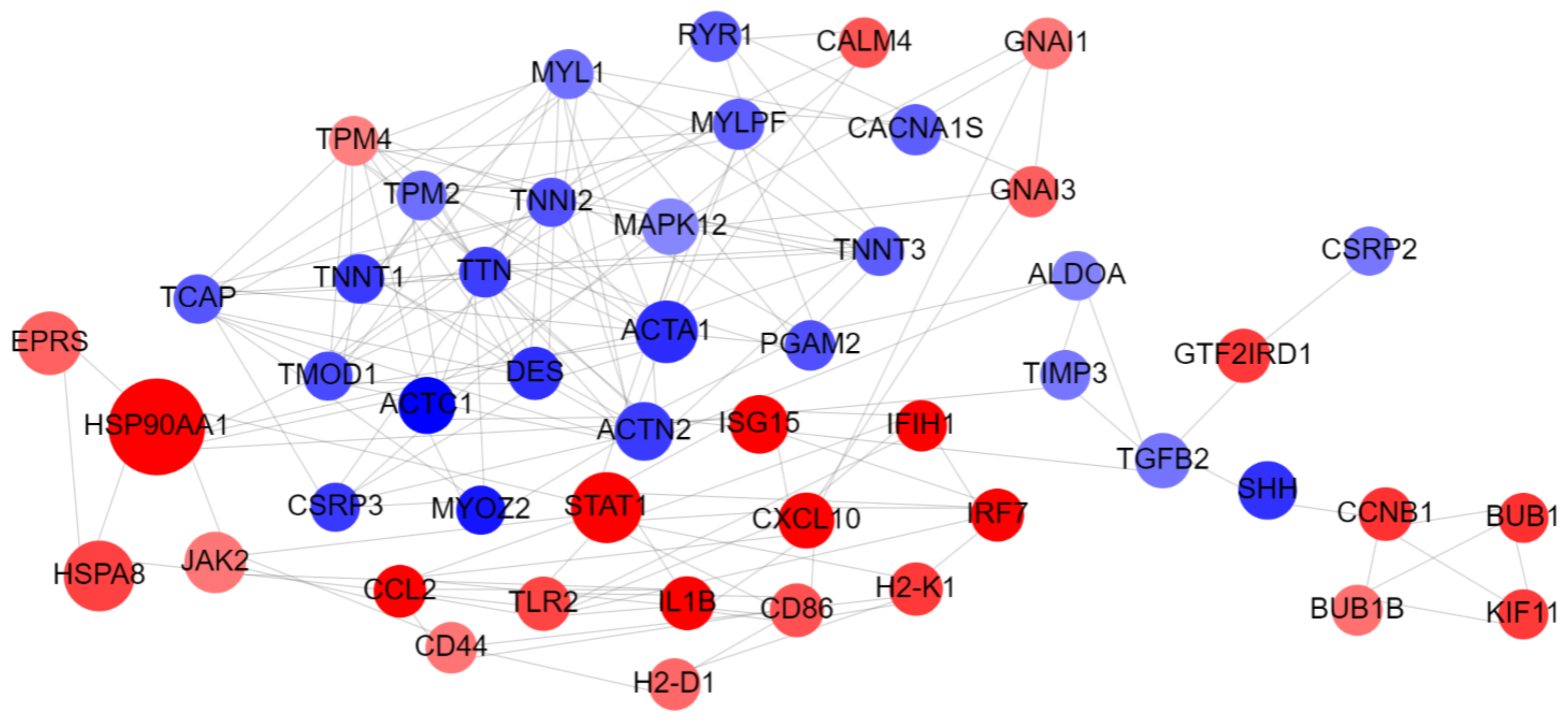
3.1. Case Study 1: GSE179385 Dataset
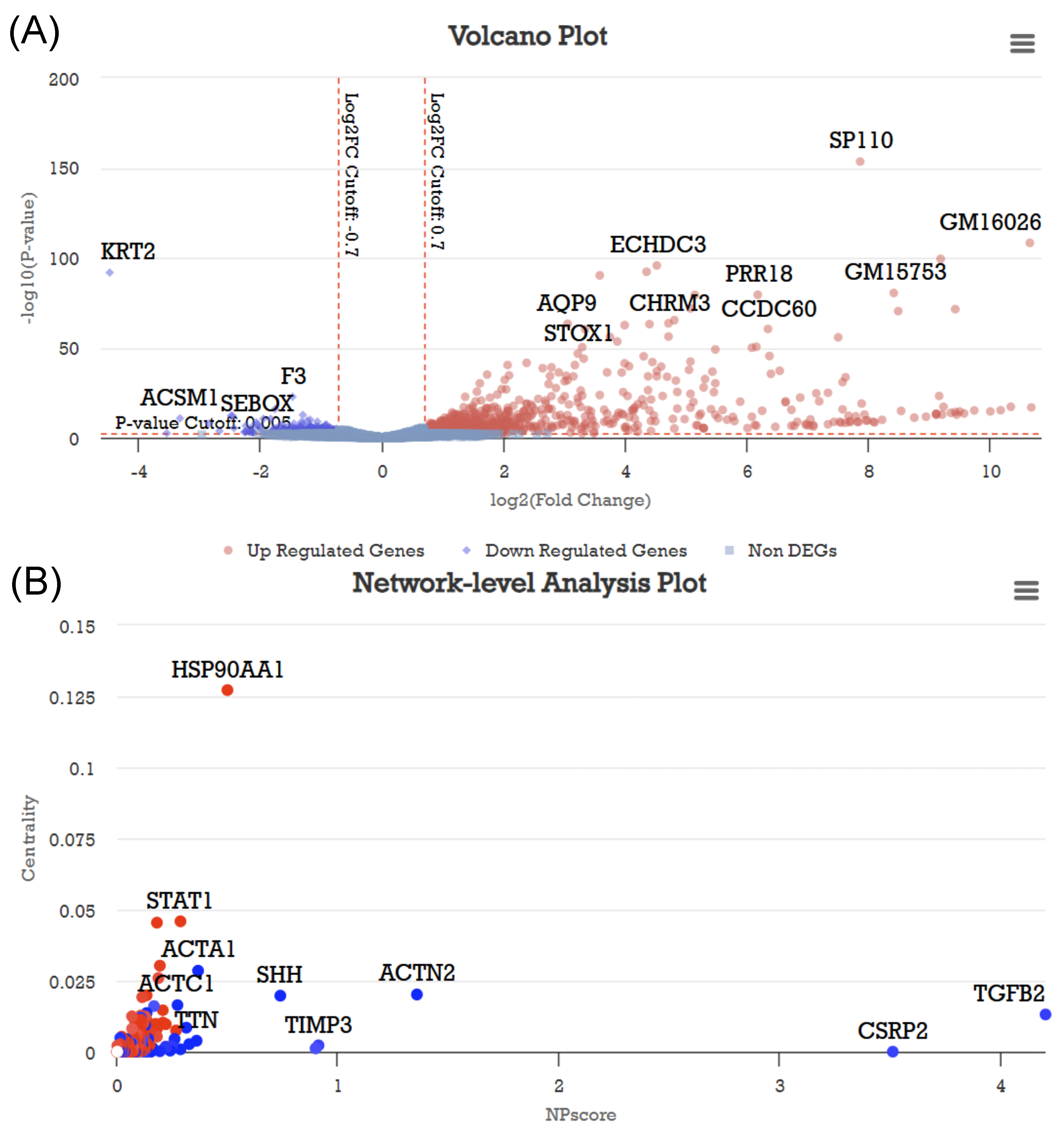
3.2. Case Study 2: GSE81082 Dataset
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 1–19. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.S.; Menotti, D.; Hungria, M.; Lopes, F.M. Temporal progress of gene expression analysis with RNA-Seq data: A review on the relationship between computational methods. Comput. Struct. Biotechnol. J. 2022. [Google Scholar] [CrossRef]
- Seyednasrollah, F.; Laiho, A.; Elo, L.L. Comparison of software packages for detecting differential expression in RNA-seq studies. Briefings Bioinform. 2015, 16, 59–70. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Z.; Zimmermann, M.T.; Bugrim, A.; Kocher, J.P. Predict drug sensitivity of cancer cells with pathway activity inference. BMC Med. Genom. 2019, 12, S5. [Google Scholar] [CrossRef]
- Allocco, D.J.; Kohane, I.S.; Butte, A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinform. 2004, 5, 18. [Google Scholar] [CrossRef]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 2016, 48, 27. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Karlebach, G.; Shamir, R. Modelling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 2008, 9, 770–780. [Google Scholar] [CrossRef]
- Gardy, J.L.; Lynn, D.J.; Brinkman, F.S.; Hancock, R.E. Enabling a systems biology approach to immunology: Focus on innate immunity. Trends Immunol. 2009, 30, 249–262. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Matys, V.; Fricke, E.; Geffers, R.; Gößling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V.; et al. TRANSFAC®: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, W.; Zhang, H.M.; Xie, G.Y.; Miao, Y.R.; Xia, M.; Guo, A.Y. hTFtarget: A comprehensive database for regulations of human transcription factors and their targets. Genom. Proteom. Bioinform. 2020, 18, 120–128. [Google Scholar] [CrossRef]
- Cowen, L.; Ideker, T.; Raphael, B.J.; Sharan, R. Network propagation: A universal amplifier of genetic associations. Nat. Rev. Genet. 2017, 18, 551–562. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, J.; Ideker, T. Classifying tumors by supervised network propagation. Bioinformatics 2018, 34, i484–i493. [Google Scholar] [CrossRef]
- Pak, M.; Jeong, D.; Moon, J.H.; Ann, H.; Hur, B.; Lee, S.; Kim, S. Network propagation for the analysis of multi-omics data. In Recent Advances in Biological Network Analysis: Comparative Network Analysis and Network Module Detection; Springer: Berlin/Heidelberg, Germany, 2021; pp. 185–217. [Google Scholar]
- Barel, G.; Herwig, R. NetCore: A network propagation approach using node coreness. Nucleic Acids Res. 2020, 48, e98. [Google Scholar] [CrossRef]
- Charmpi, K.; Chokkalingam, M.; Johnen, R.; Beyer, A. Optimizing network propagation for multi-omics data integration. PLoS Comput. Biol. 2021, 17, e1009161. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 2023, 39, btac757. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Lopes, C.T.; Fong, D.; Kucera, M.; Cheung, M.; Siper, M.C.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape. js 2023 update: A graph theory library for visualization and analysis. Bioinformatics 2023, 39, btad031. [Google Scholar] [CrossRef]
- Han, J.S.; Jeon, Y.G.; Oh, M.; Lee, G.; Nahmgoong, H.; Han, S.M.; Choi, J.; Kim, Y.Y.; Shin, K.C.; Kim, J.; et al. Adipocyte HIF2α functions as a thermostat via PKA Cα regulation in beige adipocytes. Nat. Commun. 2022, 13, 3268. [Google Scholar] [CrossRef]
- Wu, Q.; Liang, X.; Wang, K.; Lin, J.; Wang, X.; Wang, P.; Zhang, Y.; Nie, Q.; Liu, H.; Zhang, Z.; et al. Intestinal hypoxia-inducible factor 2α regulates lactate levels to shape the gut microbiome and alter thermogenesis. Cell Metab. 2021, 33, 1988–2003. [Google Scholar] [CrossRef]
- Cai, H.; Dong, L.Q.; Liu, F. Recent advances in adipose mTOR signaling and function: Therapeutic prospects. Trends Pharmacol. Sci. 2016, 37, 303–317. [Google Scholar] [CrossRef]
- Ye, Y.; Liu, H.; Zhang, F.; Hu, F. mTOR signaling in Brown and Beige adipocytes: Implications for thermogenesis and obesity. Nutr. Metab. 2019, 16, 74. [Google Scholar] [CrossRef]
- Xue, B.; Coulter, A.; Rim, J.S.; Koza, R.A.; Kozak, L.P. Transcriptional synergy and the regulation of Ucp1 during brown adipocyte induction in white fat depots. Mol. Cell. Biol. 2005, 25, 8311–8322. [Google Scholar] [CrossRef]
- Hondares, E.; Rosell, M.; Díaz-Delfín, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. In Advances in Physiology Education; American Physiological Society: Washington, DC, USA, 2006. [Google Scholar]
- Wang, X.; Dalkic, E.; Wu, M.; Chan, C. Gene module level analysis: Identification to networks and dynamics. Curr. Opin. Biotechnol. 2008, 19, 482–491. [Google Scholar] [CrossRef]
- Sanford, L.P.; Ormsby, I.; Groot, A.C.G.d.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef]
- Chimge, N.O.; Mungunsukh, O.; Ruddle, F.; Bayarsaihan, D. Expression profiling of BEN regulated genes in mouse embryonic fibroblasts. J. Exp. Zool. Part B 2007, 308, 209–224. [Google Scholar] [CrossRef]
- Makeyev, A.V.; Bayarsaihan, D. New TFII-I family target genes involved in embryonic development. Biochem. Biophys. Res. Commun. 2009, 386, 554–558. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, J.H.; Oh, M. TFNetPropX: A Web-Based Comprehensive Analysis Tool for Exploring Condition-Specific RNA-Seq Data Using Transcription Factor Network Propagation. Appl. Sci. 2023, 13, 11399. https://doi.org/10.3390/app132011399
Moon JH, Oh M. TFNetPropX: A Web-Based Comprehensive Analysis Tool for Exploring Condition-Specific RNA-Seq Data Using Transcription Factor Network Propagation. Applied Sciences. 2023; 13(20):11399. https://doi.org/10.3390/app132011399
Chicago/Turabian StyleMoon, Ji Hwan, and Minsik Oh. 2023. "TFNetPropX: A Web-Based Comprehensive Analysis Tool for Exploring Condition-Specific RNA-Seq Data Using Transcription Factor Network Propagation" Applied Sciences 13, no. 20: 11399. https://doi.org/10.3390/app132011399
APA StyleMoon, J. H., & Oh, M. (2023). TFNetPropX: A Web-Based Comprehensive Analysis Tool for Exploring Condition-Specific RNA-Seq Data Using Transcription Factor Network Propagation. Applied Sciences, 13(20), 11399. https://doi.org/10.3390/app132011399