

Emerging Insights into the Role of BDNF on Health and Disease in Periphery

 , , and
, , and
Abstract
1. Neurotrophins
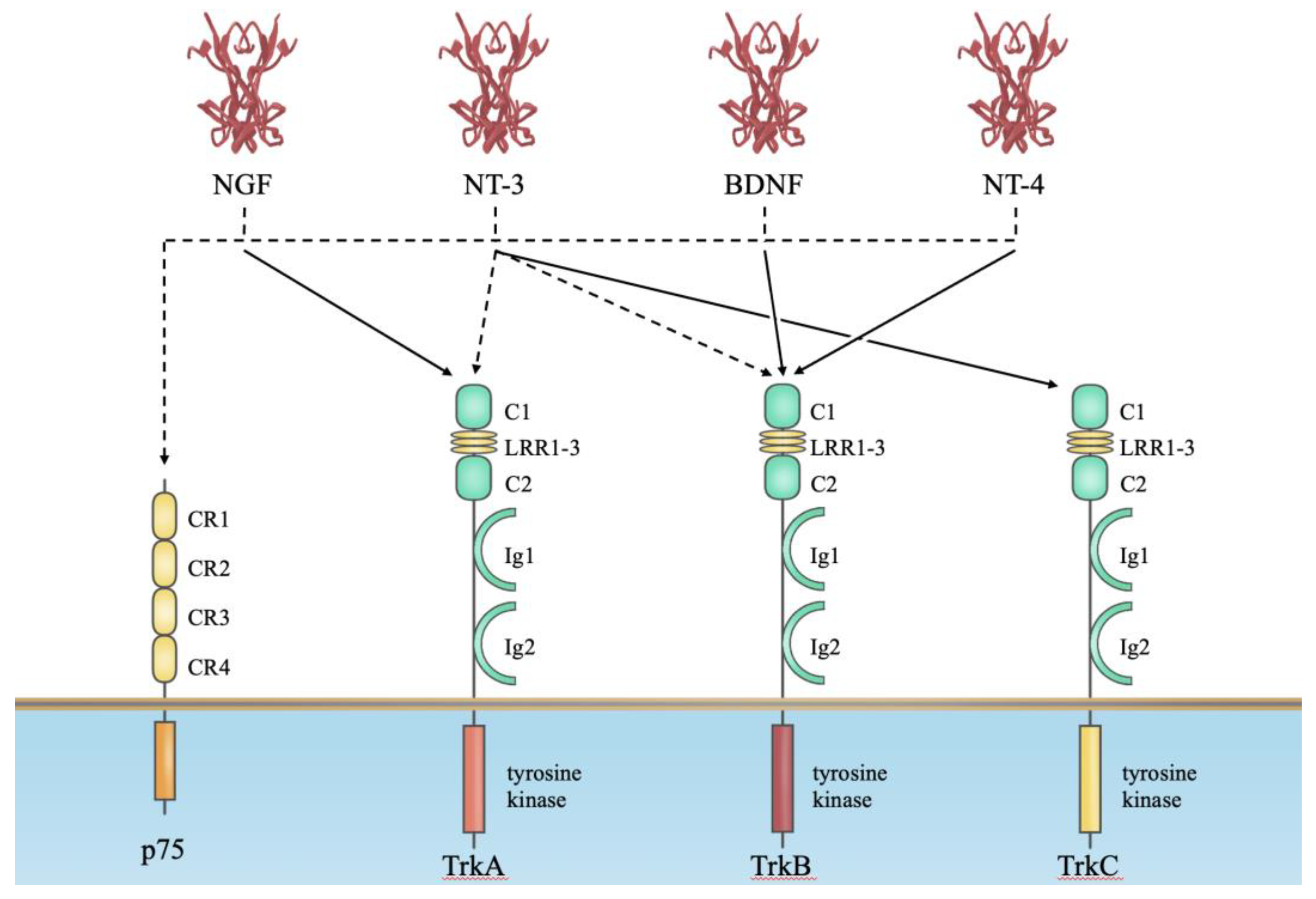
2. Neurotrophin Receptors for Their Biological Actions

3. BDNF Is a Regulator of Energy Balance, Body Weight and Food Intake
4. Role of BDNF in Insulin-Secretion Mechanism and Exercise
5. Metabolic Syndrome and Steatohepatitis
6. Animal Models of MASLD/MASH
7. BDNF Hypothesis in MASH
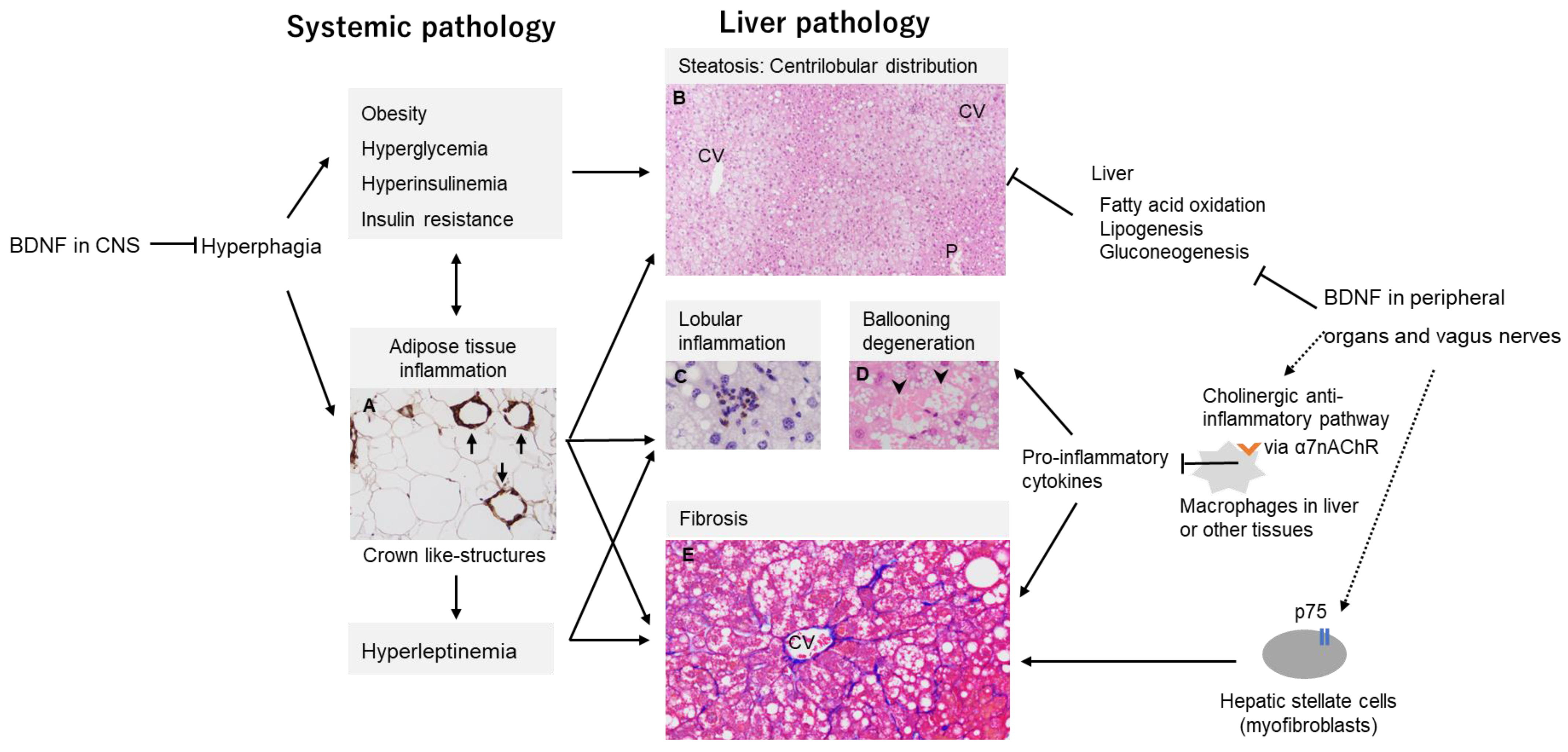
7.1. MASH Development by CNS-Mediated Effects of BDNF
7.2. Non-CNS Mediated Effects of BDNF on MASH Development
7.3. Role of Chromatin Structure and Gene Transcription Mechanism of BDNF Gene in MASH Pathogenesis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Levi-Montalcini, R. Developmental neurobiology and the natural history of nerve growth factor. Annu. Rev. Neurosci. 1982, 5, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R. The nerve growth factor and the neuroscience chess board. Prog. Brain Res. 2004, 146, 525–527. [Google Scholar] [CrossRef]
- Shelton, D.L.; Reichardt, L.F. Studies on the expression of the beta nerve growth factor (NGF) gene in the central nervous system: Level and regional distribution of NGF mRNA suggest that NGF functions as a trophic factor for several distinct populations of neurons. Proc. Natl. Acad. Sci. USA 1986, 83, 2714–2718. [Google Scholar] [CrossRef] [PubMed]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Hohn, A.; Leibrock, J.; Bailey, K.; Barde, Y.-A. Identification and characterization of a novel member of the nerve growth factor/brain-derived neurotrophic factor family. Nature 1990, 344, 339–341. [Google Scholar] [CrossRef]
- Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoenen, H.; Barde, Y.-A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature 1989, 341, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Bibel, M.; Barde, Y.-A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Meis, S.; Endres, T.; Lessmann, V. Neurotrophin signalling in amygdala-dependent cued fear learning. Cell Tissue Res. 2020, 382, 161–172. [Google Scholar] [CrossRef]
- Gonzalez, A.; Moya-Alvarado, G.; Gonzalez-Billaut, C.; Bronfman, F.C. Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton 2016, 73, 612–628. [Google Scholar] [CrossRef]
- Zagrebelsky, M.; Tacke, C.; Korte, M. BDNF signaling during the lifetime of dendritic spines. Cell Tissue Res. 2020, 382, 185–199. [Google Scholar] [CrossRef]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Le Beau, M.M.; Espinosa, R., 3rd; Ip, N.Y.; Belluscio, L.; de la Monte, S.M.; Squinto, S.; Furth, M.E.; Yancopoulos, G.D. Human and rat brain-derived neurotrophic factor and neurotrophin-3: Gene structures, distributions, and chromosomal localizations. Genomics 1991, 10, 558–568. [Google Scholar] [CrossRef]
- Ip, N.Y.; Ibáñez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Le Beau, M.M.; Espinosa, R., 3rd; Squinto, S.P.; et al. Mammalian neurotrophin-4: Structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Mizui, T.; Ishikawa, Y.; Kumanogoh, H.; Lume, M.; Matsumoto, T.; Hara, T.; Yamawaki, S.; Takahashi, M.; Shiosaka, S.; Itami, C.; et al. BDNF pro-peptide actions facilitate hippocampal LTD and are altered by the common BDNF polymorphism Val66Met. Proc. Natl. Acad. Sci. USA 2015, 112, E3067–E3074. [Google Scholar] [CrossRef]
- Mizui, T.; Hattori, K.; Ishiwata, S.; Hidese, S.; Yoshida, S.; Kunugi, H.; Kojima, M. Cerebrospinal fluid BDNF pro-peptide levels in major depressive disorder and schizophrenia. J. Psychiatr. Res. 2019, 113, 190–198. [Google Scholar] [CrossRef]
- Götz, R.; Köster, R.; Winkler, C.; Raulf, F.; Lottspeich, F.; Schartl, M.; Thoenen, H. Neurotrophin-6 is a new member of the nerve growth factor family. Nature 1994, 372, 266–269. [Google Scholar] [CrossRef]
- Ip, F.C.; Cheung, J.; Ip, N.Y. The expression profiles of neurotrophins and their receptors in rat and chicken tissues during development. Neurosci. Lett. 2001, 301, 107–110. [Google Scholar] [CrossRef]
- Thoenen, H.; Barde, Y.A. Physiology of nerve growth factor. Physiol. Rev. 1980, 60, 1284–1335. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, B.L. The many faces of p75NTR. Curr. Opin. Neurobiol. 2002, 12, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Reichardt, L.F. Trk receptors: Mediators of neurotrophin action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Tessarollo, L.; Yanpallewar, S. TrkB Truncated Isoform Receptors as Transducers and Determinants of BDNF Functions. Front. Neurosci. 2022, 16, 847572. [Google Scholar] [CrossRef]
- Escandon, E.; Soppet, D.; Rosenthal, A.; Mendoza-Ramirez, J.; Szonyi, E.; Burton, L.; Henderson, C.; Parada, L.; Nikolics, K. Regulation of neurotrophin receptor expression during embryonic and postnatal development. J. Neurosci. 1994, 14, 2054–2068. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stoilov, P.; Castren, E.; Stamm, S. Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem. Biophys. Res. Commun. 2002, 290, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, G.; Tomassoni-Ardori, F.; Babini, L.; Becker, J.; Barrick, C.; Puverel, S.; Tessarollo, L. BDNF modulates heart contraction force and long-term homeostasis through truncated TrkB.T1 receptor activation. J. Cell Biol. 2015, 210, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; O’Rahilly, S. 20 YEARS OF LEPTIN: Human disorders of leptin action. J. Endocrinol. 2014, 223, T63–T70. [Google Scholar] [CrossRef]
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578. [Google Scholar] [CrossRef]
- Richard, D. Cognitive and autonomic determinants of energy homeostasis in obesity. Nat. Rev. Endocrinol. 2015, 11, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Lapchak, P.A.; Hefti, F. BDNF and NGF treatment in lesioned rats: Effects on cholinergic function and weight gain. Neuroreport 1992, 3, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Kernie, S.G.; Liebl, D.J.; Parada, L.F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000, 19, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Lyons, W.E.; Mamounas, L.A.; Ricaurte, G.A.; Coppola, V.; Reid, S.W.; Bora, S.H.; Wihler, C.; Koliatsos, V.E.; Tessarollo, L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc. Natl. Acad. Sci. USA 1999, 96, 15239–15244. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.S.H.; Connie Hung, C.-C.; Rochford, J.; Keogh, J.; Gray, J.; Sivaramakrishnan, S.; O’Rahilly, S.; Farooqi, I.S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 2004, 7, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Yeo, G.S.H.; Cox, J.J.; Morton, J.; Adlam, A.-L.R.; Keogh, J.M.; Yanovski, J.A.; El Gharbawy, A.; Han, J.C.; Tung, Y.C.L.; et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) Gene. Diabetes 2006, 55, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Yeo, G.; Hung, C.; Keogh, J.; Clayton, P.; Banerjee, K.; McAulay, A.; O’Rahilly, S.; Farooqi, I.S. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. 2007, 31, 359–364. [Google Scholar] [CrossRef]
- Han, J.C.; Liu, Q.-R.; Jones, M.; Levinn, R.L.; Menzie, C.M.; Jefferson-George, K.S.; Adler-Wailes, D.C.; Sanford, E.L.; Lacbawan, F.L.; Uhl, G.R.; et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 2008, 359, 918–927. [Google Scholar] [CrossRef]
- Xu, B.; Goulding, E.H.; Zang, K.; Cepoi, D.; Cone, R.D.; Jones, K.R.; Tecott, L.H.; Reichardt, L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003, 6, 736–742. [Google Scholar] [CrossRef]
- Unger, T.J.; Calderon, G.A.; Bradley, L.C.; Sena-Esteves, M.; Rios, M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J. Neurosci. 2007, 27, 14265–14274. [Google Scholar] [CrossRef]
- Tran, P.V.; Akana, S.F.; Malkovska, I.; Dallman, M.F.; Parada, L.F.; Ingraham, H.A. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J. Comp. Neurol. 2006, 498, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xie, X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 2016, 17, 282–292. [Google Scholar] [CrossRef]
- Kahali, B.; Halligan, B.; Speliotes, E.K. Insights from genome-wide association analyses of nonalcoholic fatty liver disease. Semin. Liver Dis. 2015, 35, 375–391. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Voss, T.C.; Hager, G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014, 15, 69–81. [Google Scholar] [CrossRef]
- Soshnev, A.A.; Josefowicz, S.Z.; Allis, C.D. Greater than the sum of parts: Complexity of the dynamic epigenome. Mol. Cell 2016, 62, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, G.; Hong, Z.; Tomassoni-Ardori, F.; Barella, L.F.; Becker, J.; Barrick, C.; Swing, D.; Yanpallewar, S.; Croix, B.S.; Wess, J.; et al. Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 2020, 11, 1950. [Google Scholar] [CrossRef]
- Wang, R.-R.; Pan, R.; Zhang, W.; Fu, J.; Lin, J.D.; Meng, Z.-X. The SWI/SNF chromatin-remodeling factors BAF60a, b, and c in nutrient signaling and metabolic control. Protein Cell 2018, 9, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Ichimura-Shimizu, M.; Kojima, M.; Suzuki, S.; Miyata, M.; Osaki, Y.; Matsui, K.; Mizui, T.; Tsuneyama, K. Brain-derived neurotrophic factor knock-out mice develop non-alcoholic steatohepatitis. J. Pathol. 2023, 261, 465–476. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Koch, L.K.; Yeh, M.M. Nonalcoholic fatty liver disease (NAFLD): Diagnosis, pitfalls, and staging. Ann. Diagn. Pathol. 2018, 37, 83–90. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: Revisited after a decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Du Plessis, J.; Van Pelt, J.; Korf, H.; Mathieu, C.; Van Der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology 2015, 149, 635–648.e14. [Google Scholar] [CrossRef]
- Duval, C.; Thissen, U.; Keshtkar, S.; Accart, B.; Stienstra, R.; Boekschoten, M.V.; Roskams, T.; Kersten, S.; Muller, M. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57Bl/6 Mice. Diabetes 2010, 59, 3181–3191. [Google Scholar] [CrossRef]
- Albillos, A.; De Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Horn, C.L.; Morales, A.L.; Savard, C.; Farrell, G.C.; Ioannou, G.N. Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol. Commun. 2022, 6, 12–35. [Google Scholar] [CrossRef]
- Teng, T.; Qiu, S.; Zhao, Y.; Zhao, S.; Sun, D.; Hou, L.; Li, Y.; Zhou, K.; Yu, X.; Yang, C.; et al. Pathogenesis and therapeutic strategies related to non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2022, 23, 7841. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M. Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 2008, 23, 1635–1648. [Google Scholar] [CrossRef]
- Pickens, M.K.; Yan, J.S.; Ng, R.K.; Ogata, H.; Grenert, J.P.; Beysen, C.; Turner, S.M.; Maher, J.J. Dietary sucrose is essential to the development of liver injury in the methionine-choline-deficient model of steatohepatitis. J. Lipid Res. 2009, 50, 2072–2082. [Google Scholar] [CrossRef]
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653. [Google Scholar] [CrossRef]
- Yao, Z.M.; Vance, D.E. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 1988, 263, 2998–3004. [Google Scholar] [CrossRef]
- Recena Aydos, L.; Aparecida do Amaral, L.; Serafim de Souza, R.; Jacobowski, A.C.; Freitas dos Santos, E.; Rodrigues Macedo, M.L. Nonalcoholic fatty liver disease induced by high-fat diet in C57bl/6 models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Vergnes, L.; Phan, J.; Strauss, M.; Tafuri, S.; Reue, K. Cholesterol and cholate components of an atherogenic diet induce distinct stages of hepatic inflammatory gene expression. J. Biol. Chem. 2003, 278, 42774–42784. [Google Scholar] [CrossRef]
- Ichimura, M.; Kawase, M.; Masuzumi, M.; Sakaki, M.; Nagata, Y.; Tanaka, K.; Suruga, K.; Tamaru, S.; Kato, S.; Tsuneyama, K.; et al. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague–Dawley rats. Hepatol. Res. 2015, 45, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, M.; Masuzumi, M.; Kawase, M.; Sakaki, M.; Tamaru, S.; Nagata, Y.; Tanaka, K.; Suruga, K.; Tsuneyama, K.; Matsuda, S.; et al. A diet-induced Sprague–Dawley rat model of nonalcoholic steatohepatitis-related cirrhosis. J. Nutr. Biochem. 2017, 40, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. J. Hepatol. 2007, 46, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Ichimura-Shimizu, M.; Omagari, K.; Yamashita, M.; Tsuneyama, K. Development of a novel mouse model of diet-induced nonalcoholic steatohepatitis–related progressive bridging fibrosis. Biosci. Biotechnol. Biochem. 2021, 85, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Makiuchi, N.; Takano, S.; Tada, Y.; Kasai, K.; Igarashi, N.; Kani, K.; Kato, M.; Goto, K.; Matsuura, Y.; Ichimura-Shimizu, M.; et al. Dynamics of liver macrophage subsets in a novel mouse model of non-alcoholic steatohepatitis using C57BL/6 Mice. Biomedicines 2023, 11, 2659. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Kasai, K.; Makiuchi, N.; Igarashi, N.; Kani, K.; Takano, S.; Honda, H.; Yanagibashi, T.; Watanabe, Y.; Usui-Kawanishi, F.; et al. Roles of macrophages in advanced liver fibrosis, identified using a newly established mouse model of diet-induced non-alcoholic steatohepatitis. Int. J. Mol. Sci. 2022, 23, 13251. [Google Scholar] [CrossRef] [PubMed]
- Ichimura-Shimizu, M.; Tsuchiyama, Y.; Morimoto, Y.; Matsumoto, M.; Kobayashi, T.; Sumida, S.; Kakimoto, T.; Oya, T.; Ogawa, H.; Yamashita, M.; et al. A novel mouse model of nonalcoholic steatohepatitis suggests that liver fibrosis initiates around lipid-laden macrophages. Am. J. Pathol. 2022, 192, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, A.H.; Clinton, S.K.; Iiyama, K.; Connelly, P.W.; Libby, P.; Cybulsky, M.I. Hyperlipidemia and atherosclerotic lesion development in LDL receptor–deficient mice fed defined semipurified diets with and without cholate. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, I.A.; Farrell, G.C.; Schriemer, R.; Robertson, G.R. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 2002, 37, 206–213. [Google Scholar] [CrossRef]
- Ikejima, K.; Takei, Y.; Honda, H.; Hirose, M.; Yoshikawa, M.; Zhang, Y.-J.; Lang, T.; Fukuda, T.; Yamashina, S.; Kitamura, T.; et al. Leptin receptor–mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002, 122, 1399–1410. [Google Scholar] [CrossRef]
- Itoh, M.; Suganami, T.; Nakagawa, N.; Tanaka, M.; Yamamoto, Y.; Kamei, Y.; Terai, S.; Sakaida, I.; Ogawa, Y. Melanocortin 4 receptor–deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol. 2011, 179, 2454–2463. [Google Scholar] [CrossRef]
- Itoh, M.; Kato, H.; Suganami, T.; Konuma, K.; Marumoto, Y.; Terai, S.; Sakugawa, H.; Kanai, S.; Hamaguchi, M.; Fukaishi, T.; et al. Hepatic crown-like structure: A unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS ONE 2013, 8, e82163. [Google Scholar] [CrossRef]
- Larter, C.Z.; Chitturi, S.; Heydet, D.; Farrell, G.C. A fresh look at NASH pathogenesis. Part 1: The metabolic movers. J. Gastroenterol. Hepatol. 2010, 25, 672–690. [Google Scholar] [CrossRef]
- Liao, G.-Y.; An, J.J.; Gharami, K.; Waterhouse, E.G.; Vanevski, F.; Jones, K.R.; Xu, B. Dendritically targeted Bdnf mRNA is essential for energy balance and response to leptin. Nat. Med. 2012, 18, 564–571. [Google Scholar] [CrossRef]
- Wu, L.; Gao, X.; Guo, Q.; Li, J.; Yao, J.; Yan, K.; Xu, Y.; Jiang, X.; Ye, D.; Guo, J. The role of neutrophils in innate immunity-driven nonalcoholic steatohepatitis: Lessons learned and future promise. Hepatol. Int. 2020, 14, 652–666. [Google Scholar] [CrossRef]
- Zang, S.; Wang, L.; Ma, X.; Zhu, G.; Zhuang, Z.; Xun, Y.; Zhao, F.; Yang, W.; Liu, J.; Luo, Y.; et al. Neutrophils play a crucial role in the early stage of nonalcoholic steatohepatitis via neutrophil elastase in mice. Cell Biochem. Biophys. 2015, 73, 479–487. [Google Scholar] [CrossRef]
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 86–95. [Google Scholar] [CrossRef]
- Di Rosa, M.C.; Zimbone, S.; Saab, M.W.; Tomasello, M.F. The pleiotropic potential of BDNF beyond neurons: Implication for a healthy mind in a healthy body. Life 2021, 11, 1256. [Google Scholar] [CrossRef]
- Matthews, V.B.; Åström, M.-B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef]
- Severinsen, M.C.K.; Pedersen, B.K. Muscle–organ crosstalk: The emerging roles of myokines. Endocr. Rev. 2020, 41, 594–609. [Google Scholar] [CrossRef]
- Rentería, I.; García-Suárez, P.C.; Fry, A.C.; Moncada-Jiménez, J.; Machado-Parra, J.P.; Antunes, B.M.; Jiménez-Maldonado, A. The molecular effects of BDNF synthesis on skeletal muscle: A mini-review. Front. Physiol. 2022, 13, 934714. [Google Scholar] [CrossRef]
- Meek, T.H.; Wisse, B.E.; Thaler, J.P.; Guyenet, S.J.; Matsen, M.E.; Fischer, J.D.; Taborsky, G.J., Jr.; Schwartz, M.W.; Morton, G.J. BDNF action in the brain attenuates diabetic hyperglycemia via insulin-independent inhibition of hepatic glucose production. Diabetes 2013, 62, 1512–1518. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tsuchida, A.; Itakura, Y.; Nonomura, T.; Ono, M.; Hirota, F.; Inoue, T.; Nakayama, C.; Taiji, M.; Noguchi, H. Brain-derived neurotrophic factor regulates glucose metabolism by modulating energy balance in diabetic mice. Diabetes 2000, 49, 436–444. [Google Scholar] [CrossRef]
- Genzer, Y.; Chapnik, N.; Froy, O. Effect of brain-derived neurotrophic factor (BDNF) on hepatocyte metabolism. Int. J. Biochem. Cell Biol. 2017, 88, 69–74. [Google Scholar] [CrossRef]
- Nishio, T.; Taura, K.; Iwaisako, K.; Koyama, Y.; Tanabe, K.; Yamamoto, G.; Okuda, Y.; Ikeno, Y.; Yoshino, K.; Kasai, Y.; et al. Hepatic vagus nerve regulates Kupffer cell activation via α7 nicotinic acetylcholine receptor in nonalcoholic steatohepatitis. J. Gastroenterol. 2017, 52, 965–976. [Google Scholar] [CrossRef]
- Maurer, S.V.; Williams, C.L. The cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Kimura, K.; Inaba, Y.; Watanabe, H.; Matsukawa, T.; Matsumoto, M.; Inoue, H. Nicotinic alpha-7 acetylcholine receptor deficiency exacerbates hepatic inflammation and fibrosis in a mouse model of non-alcoholic steatohepatitis. J. Diabetes Investig. 2019, 10, 659–666. [Google Scholar] [CrossRef]
- Massey, K.A.; Zago, W.M.; Berg, D.K. BDNF up-regulates α7 nicotinic acetylcholine receptor levels on subpopulations of hippocampal interneurons. Mol. Cell. Neurosci. 2006, 33, 381–388. [Google Scholar] [CrossRef]
- Kawai, H.; Zago, W.; Berg, D.K. Nicotinic α7 Receptor Clusters on Hippocampal GABAergic Neurons: Regulation by Synaptic Activity and Neurotrophins. J. Neurosci. 2002, 22, 7903–7912. [Google Scholar] [CrossRef]
- Biddinger, J.E.; Fox, E.A. Reduced intestinal brain-derived neurotrophic factor increases vagal sensory innervation of the intestine and enhances satiation. J. Neurosci. 2014, 34, 10379–10393. [Google Scholar] [CrossRef]
- Passino, M.A.; Adams, R.A.; Sikorski, S.L.; Akassoglou, K. Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science 2007, 315, 1853–1856. [Google Scholar] [CrossRef]
- Teillon, S.; Calderon, G.A.; Rios, M. Diminished diet-induced hyperglycemia and dyslipidemia and enhanced expression of PPARα and FGF21 in mice with hepatic ablation of brain-derived neurotropic factor. J. Endocrinol. 2010, 205, 37–47. [Google Scholar] [CrossRef][Green Version]
- Clapper, J.R.; Hendricks, M.D.; Gu, G.; Wittmer, C.; Dolman, C.S.; Herich, J.; Athanacio, J.; Villescaz, C.; Ghosh, S.S.; Heilig, J.S.; et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, G483–G495. [Google Scholar] [CrossRef]
- Xiong, X.; Wang, Q.; Wang, S.; Zhang, J.; Liu, T.; Guo, L.; Yu, Y.; Lin, J.D. Mapping the molecular signatures of diet-induced NASH and its regulation by the hepatokine Tsukushi. Mol. Metab. 2019, 20, 128–137. [Google Scholar] [CrossRef]
- Lefebvre, P.; Lalloyer, F.; Baugé, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. J. Clin. Investig. 2017, 2, e92264. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, T.; Mi, L.; Kuang, H.; Xiong, X.; Chen, Z.; Li, S.; Lin, J.D. hnRNPU/TrkB defines a chromatin accessibility checkpoint for liver injury and nonalcoholic steatohepatitis pathogenesis. Hepatology 2020, 71, 1228–1246. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Model | Metabolic Phenotype | Liver Histology | |||||
|---|---|---|---|---|---|---|---|
| Obesity | Insulin Resistance | Adipose Tissue Inflammation | Dyslipidemia | Steatosis | Steato-Hepatitis | Fibrosis | |
| Methionine and choline deficient diet | No (weight loss) | No | No (decreased adiposity) | No | Yes | Yes | Yes |
| HFD | Yes | Yes | Yes | Yes | Yes | Yes (mild) | Yes (mild) |
| HFD with cholesterol and cholate (iHFC diet) | Yes (depends on cholate-dose) | Yes | Yes | Yes | Yes | Yes | Yes |
| ob/ob mice | Yes | Yes | Yes | Yes | Yes | No (does not develop spontaneously) | No (resistant to fibrosis) |
| db/db mice | Yes | Yes | Yes | Yes | Yes | No (does not develop spontaneously) | No (does not develop spontaneously) |
| Melanocortin 4 receptor–deficient mice | Yes | Yes | Yes | Yes | Yes | Yes | Yes (under the HFD feeding) |
| BDNF knockout (+/−) mice | Yes | Yes | Yes | No | Yes | Yes | Yes |
| proBDNF knock-in mice | Yes | Yes | Yes | No | Yes | Yes | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ichimura-Shimizu, M.; Kurrey, K.; Miyata, M.; Dezawa, T.; Tsuneyama, K.; Kojima, M. Emerging Insights into the Role of BDNF on Health and Disease in Periphery. Biomolecules 2024, 14, 444. https://doi.org/10.3390/biom14040444
Ichimura-Shimizu M, Kurrey K, Miyata M, Dezawa T, Tsuneyama K, Kojima M. Emerging Insights into the Role of BDNF on Health and Disease in Periphery. Biomolecules. 2024; 14(4):444. https://doi.org/10.3390/biom14040444
Chicago/Turabian StyleIchimura-Shimizu, Mayuko, Khuleshwari Kurrey, Misaki Miyata, Takuya Dezawa, Koichi Tsuneyama, and Masami Kojima. 2024. "Emerging Insights into the Role of BDNF on Health and Disease in Periphery" Biomolecules 14, no. 4: 444. https://doi.org/10.3390/biom14040444
APA StyleIchimura-Shimizu, M., Kurrey, K., Miyata, M., Dezawa, T., Tsuneyama, K., & Kojima, M. (2024). Emerging Insights into the Role of BDNF on Health and Disease in Periphery. Biomolecules, 14(4), 444. https://doi.org/10.3390/biom14040444