
Exploring the Antiangiogenic and Anti-Inflammatory Potential of Homoisoflavonoids: Target Identification Using Biotin Probes
 ,
,  and
and
Abstract
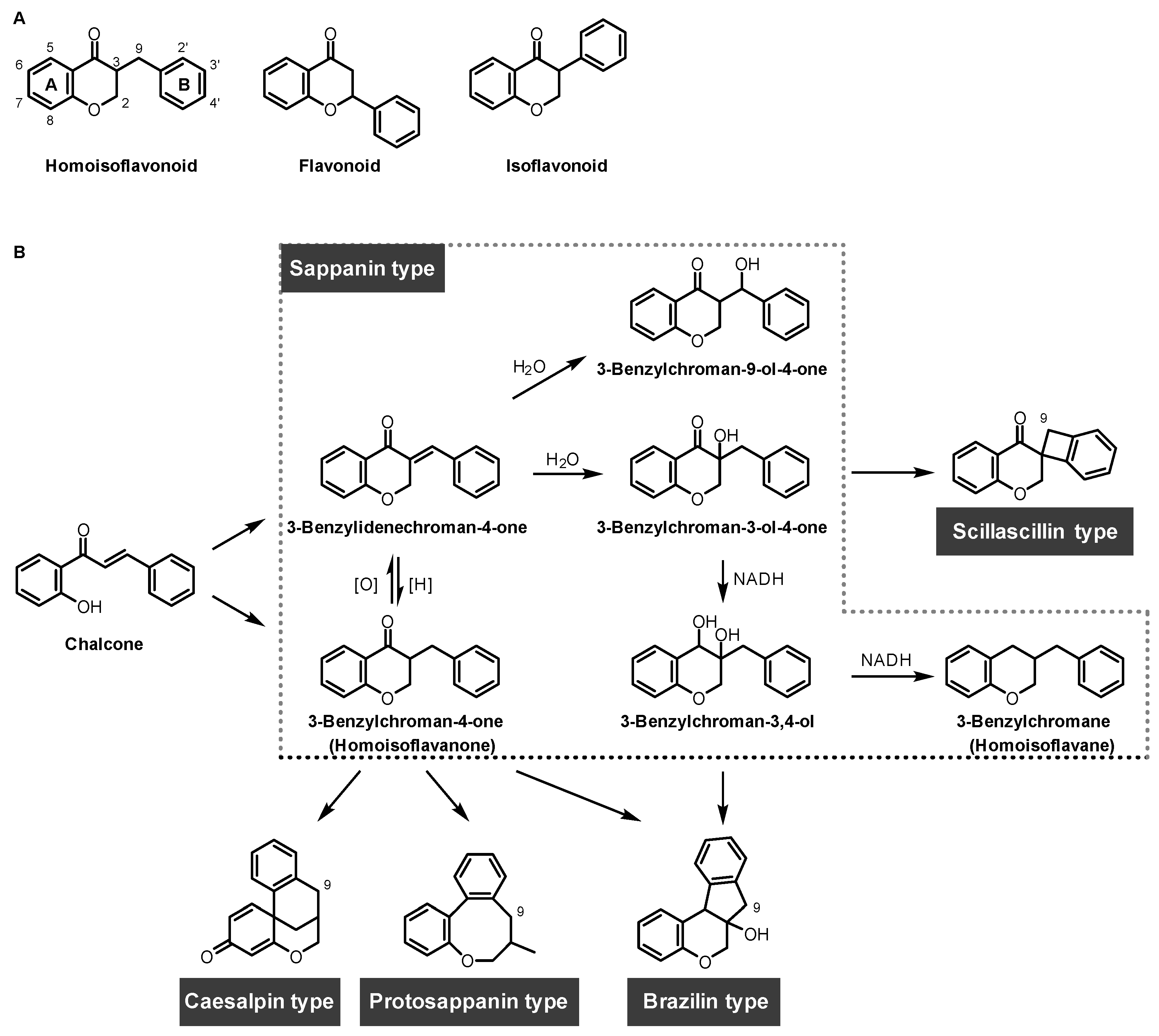
1. Introduction
2. Applications of Homoisoflavonoid Probes in Target Identification
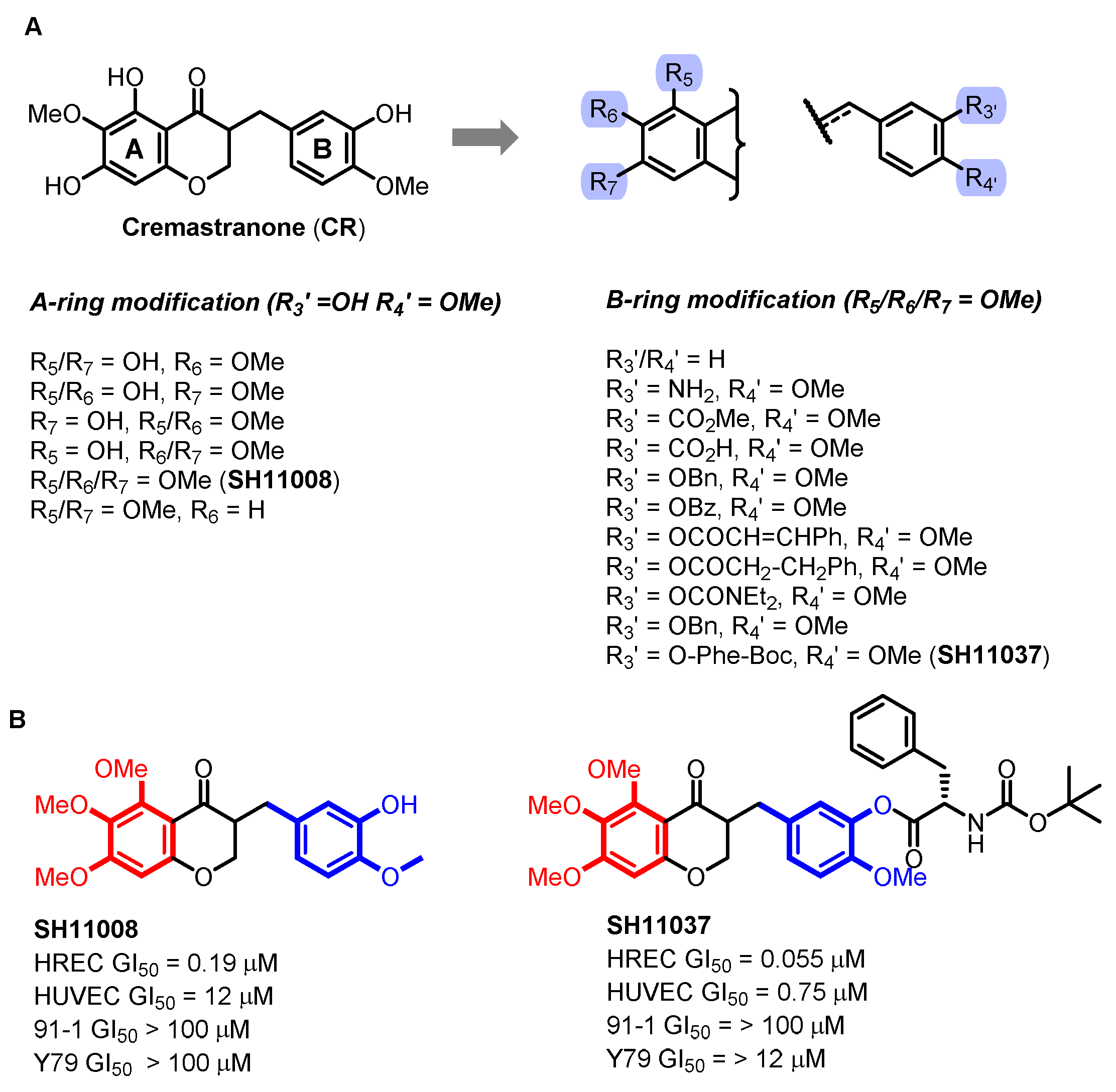
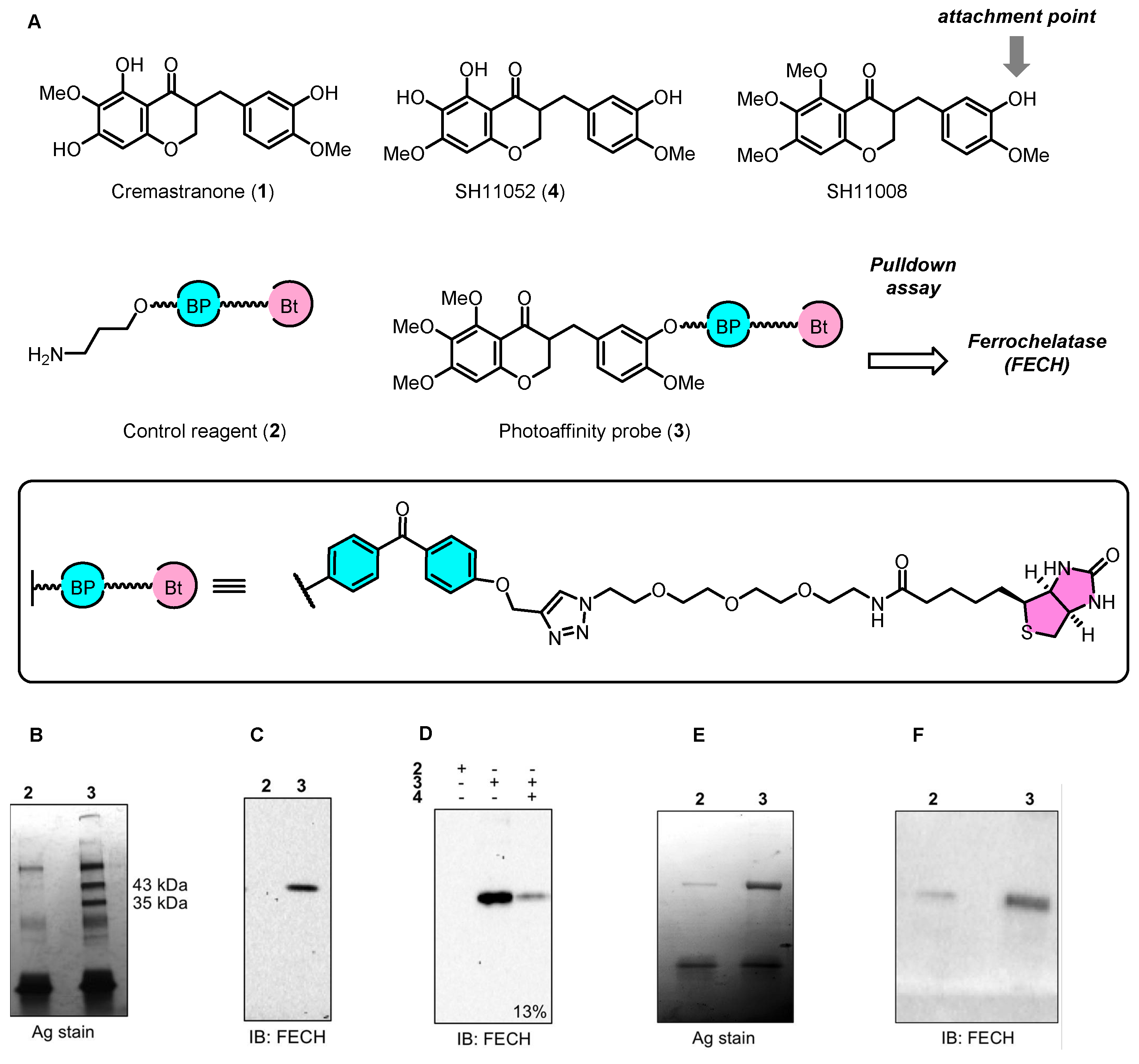
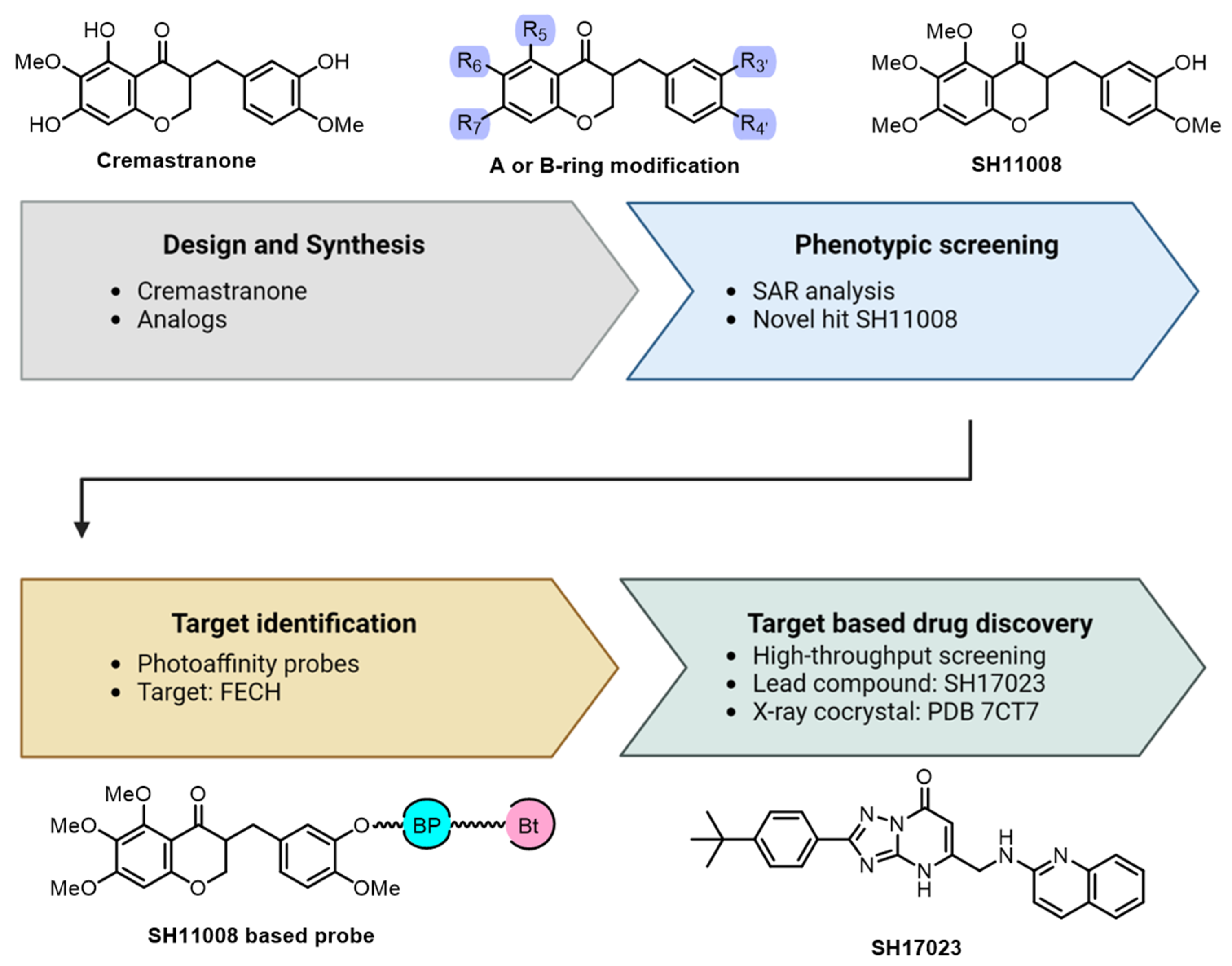
2.1. Cremastranone Inhibited Ocular Neovascularization by Targeting FECH
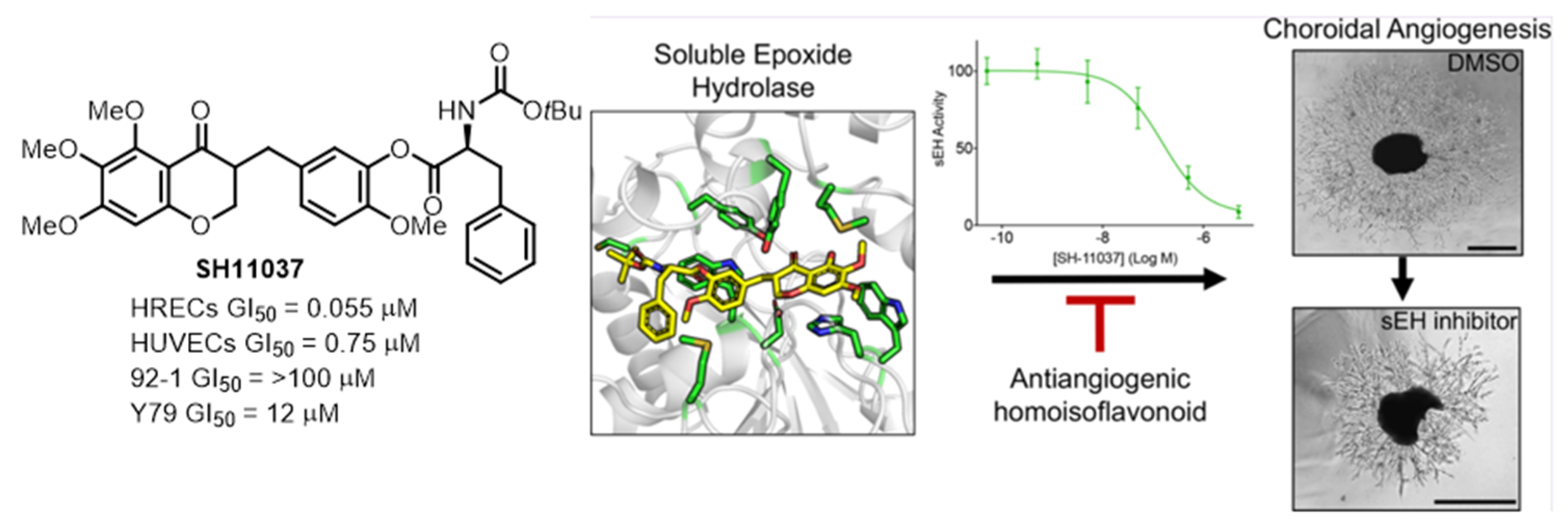
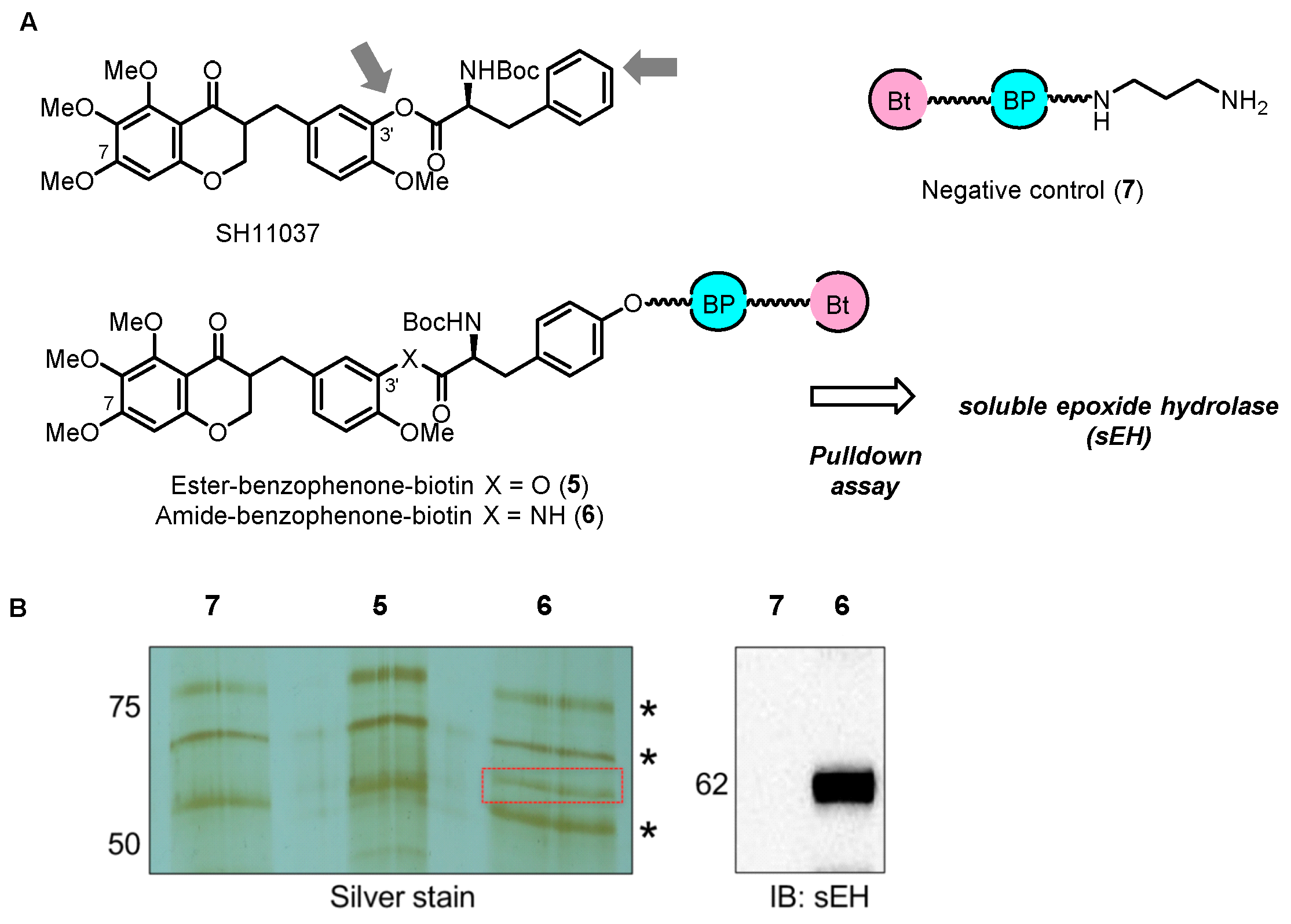
2.2. Chemical Proteomics Identified sEH as a Therapeutic Target for Ocular Neovascularization
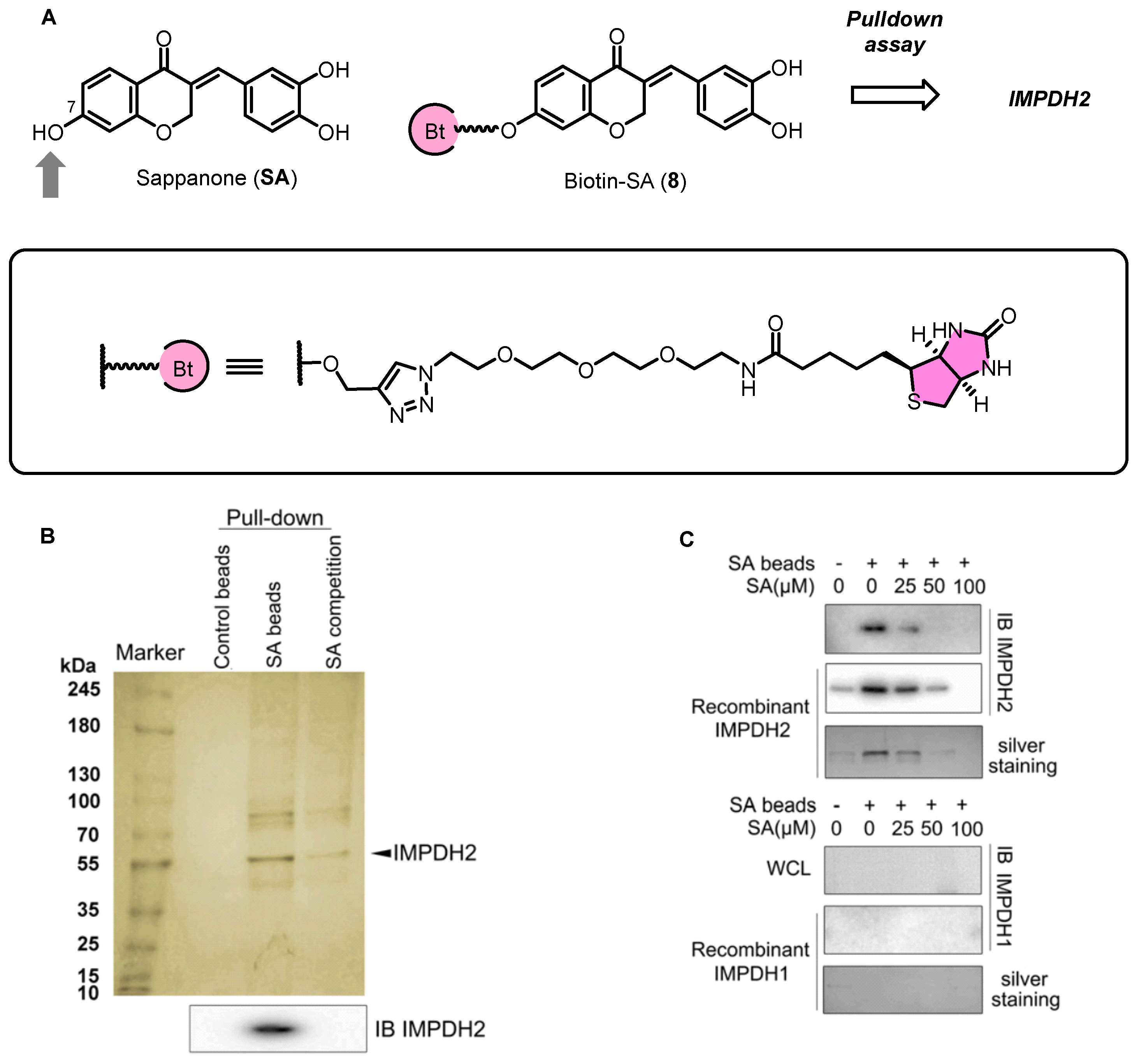
2.3. Sappanone A Inhibited IMPDH for Anti-Inflamatory Effects
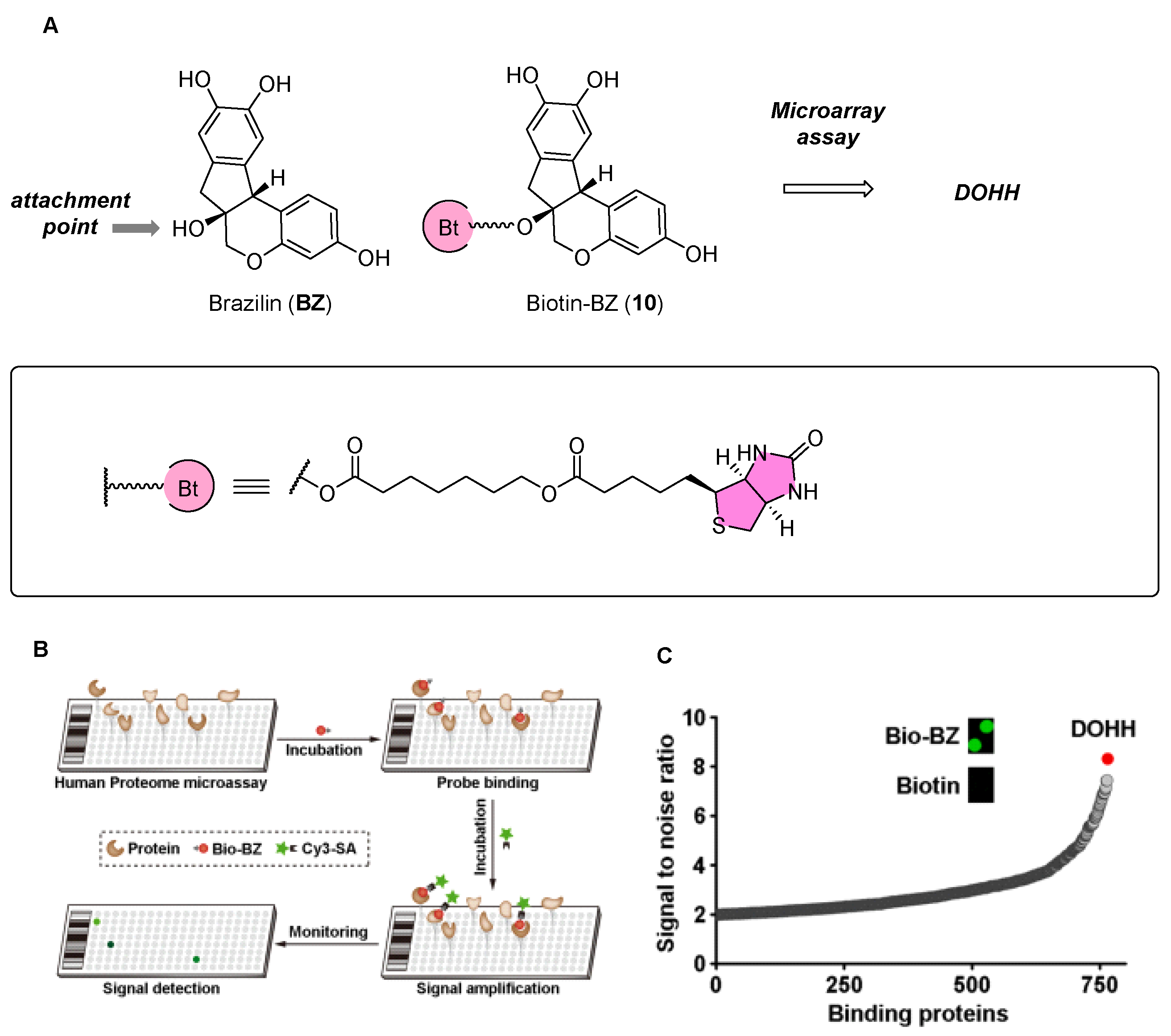
2.4. Brazilin Allosteric Activate DOHH for Neuroprotection in Ischemic Stroke
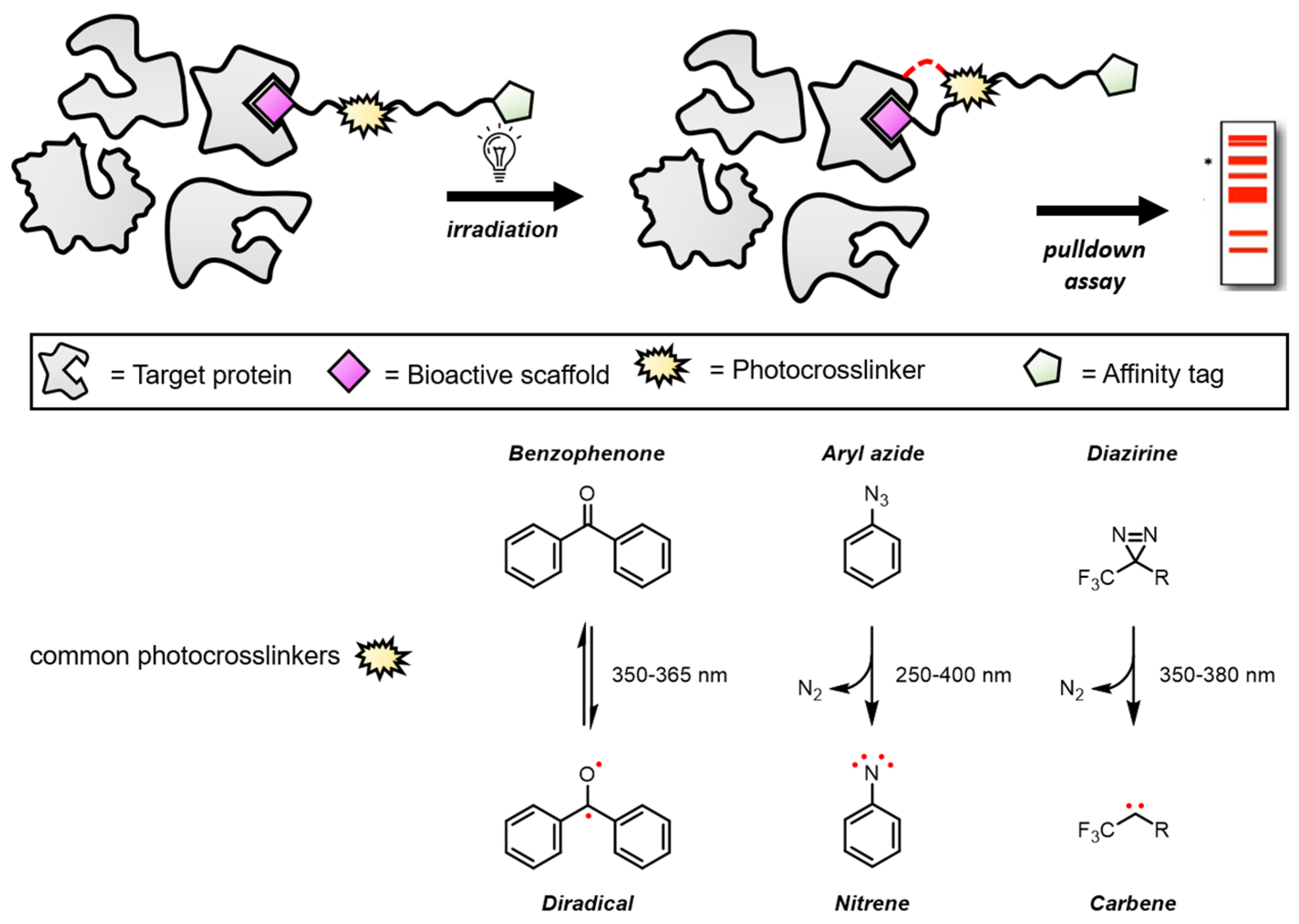
2.5. Emerging Techniques for Target Identification beyond PAL
3. Conclusions
Funding
Conflicts of Interest
References
- Bhukta, S.; Gopinath, P.; Dandela, R. Target identification of anticancer natural products using a chemical proteomics approach. RSC Adv. 2021, 11, 27950–27964. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Li, C.; Chen, P.; Yang, H. An update of label-free protein target identification methods for natural active products. Theranostics 2022, 12, 1829–1854. [Google Scholar] [CrossRef] [PubMed]
- Govindaraju, R.; Govindaraju, S.; Yun, K.; Kim, J. Fluorescent-Based Neurotransmitter Sensors: Present and Future Perspectives. Biosensors 2023, 13, 1008. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Sieber, S.A. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33, 681–708. [Google Scholar] [CrossRef] [PubMed]
- Terstappen, G.C.; Schlüpen, C.; Raggiaschi, R.; Gaviraghi, G. Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discov. 2007, 6, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Nueda, A.; Lee, J.; Schenone, M.; Prunotto, M.; Mercola, M. Phenotypic drug discovery: Recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 2022, 21, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.-Y.; Cho, S.M.; Kwon, H.J. Approaches for discovering novel bioactive small molecules targeting autophagy. Expert. Opin. Drug Discov. 2017, 12, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Komura, H.; Watanabe, R.; Mizuguchi, K. The Trends and Future Prospective of In silico Models from the Viewpoint of ADME Evaluation in Drug Discovery. Pharmaceutics 2023, 15, 2319. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; Lyu, A.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, gkae236. [Google Scholar] [CrossRef]
- Paula, C.-R.; Jose, L.-B.; Nereida, R.-F.; Francisco, C.; Francisco, J.N.; Adrian, C.; Victor, M.; Alejandro, P.; Carlos, F.-L. A review on machine learning approaches and trends in drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 4538–4558. [Google Scholar] [CrossRef]
- Corson, T.W.; Crews, C.M. Molecular understanding and modern application of traditional medicines: Triumphs and trials. Cell 2007, 130, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef]
- Van Vleet, T.R.; Liguori, M.J.; Lynch III, J.J.; Rao, M.; Warder, S. Screening strategies and methods for better off-target liability prediction and identification of small-molecule pharmaceuticals. SLAS Discov. 2019, 24, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.-Y.; Corson, T.W. Small molecule target identification using photo-affinity chromatography. Methods Enzymol. 2019, 622, 347–374. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Ma, N.; Tian, J.; Shao, Y.; Wong, Y.K.; Liang, Z.; Zou, C.; Wang, J. Target identification of natural medicine with chemical proteomics approach: Probe synthesis, target fishing and protein identification. Sig. Transduct. Target. Ther. 2020, 5, 72. [Google Scholar] [CrossRef] [PubMed]
- Murale, D.P.; Hong, S.C.; Haque, M.M.; Lee, J.-S. Photo-affinity labeling (PAL) in chemical proteomics: A handy tool to investigate protein-protein interactions (PPIs). Proteome Sci. 2017, 15, 14. [Google Scholar] [CrossRef]
- Abegaz, B.M.; Mutanyatta-Comar, J.; Nindi, M. Naturally occurring homoisoflavonoids: Phytochemistry, biological activities and synthesis. Nat. Prod. Commun. 2007, 2, 475–498. [Google Scholar] [CrossRef]
- Lin, L.-G.; Liu, Q.-Y.; Ye, Y. Naturally occurring homoisoflavonoids and their pharmacological activities. Planta Med. 2014, 80, 1053–1066. [Google Scholar] [CrossRef]
- Mulholland, D.A.; Schwikkard, S.L.; Crouch, N.R. The chemistry and biological activity of the Hyacinthaceae. Nat. Prod. Rep. 2013, 30, 1165–1210. [Google Scholar] [CrossRef]
- Castelli, M.V.; López, S.N. Homoisoflavonoids: Occurrence, biosynthesis, and biological activity. Stud. Nat. Prod. Chem. 2017, 54, 315–354. [Google Scholar] [CrossRef]
- du Toit, K.; Drewes, S.E.; Bodenstein, J. The chemical structures, plant origins, ethnobotany and biological activities of homoisoflavanones. Nat. Prod. Res. 2010, 24, 457–490. [Google Scholar] [CrossRef] [PubMed]
- Schwikkard, S.; Whitmore, H.; Sishtla, K.; Sulaiman, R.S.; Shetty, T.; Basavarajappa, H.D.; Waller, C.; Alqahtani, A.; Frankemoelle, L.; Chapman, A.; et al. The Antiangiogenic Activity of Naturally Occurring and Synthetic Homoisoflavonoids from the Hyacinthaceae (sensu APGII). J. Nat. Prod. 2019, 82, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Griffin, E.; Jefford, H.; Schwikkard, S.; Corson, T.; Mulholland, D. Synthesis of Derivatised Homoisoflavonoids to Target Ocular Angiogenesis. Planta Med. 2022, 88, 1523. [Google Scholar] [CrossRef]
- Choi, Y.; Park, S.; Lee, S.; Shin, H.-E.; Kwon, S.; Choi, J.-K.; Lee, M.-H.; Seo, S.-Y.; Lee, Y. Cremastranone-Derived Homoisoflavanes Suppress the Growth of Breast Cancer Cells via Cell Cycle Arrest and Caspase-Independent Cell Death. Biomol. Ther. 2023, 31, 526. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Z.; Wang, Y.-L.; Che, H.-J.; Jia, Y.-H.; Wang, H.-F.; Zuo, L.-F.; Yang, K.; Li, T.-T.; Wang, J.-X. Sappanone A: A natural PDE4 inhibitor with dual anti-inflammatory and antioxidant activities from the heartwood of Caesalpinia sappan L. J. Ethnopharmacol. 2023, 304, 116020. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Kim, J.H.; Lee, J.; Kim, S.N.; Kwon, H.J. Anti-angiogenic activity of a homoisoflavanone from Cremastra appendiculata. Planta Med. 2004, 70, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, K.H.; Kim, J.H.; Yu, Y.S.; Kim, Y.-M.; Kim, K.-W.; Kwon, H.J. Homoisoflavanone inhibits retinal neovascularization through cell cycle arrest with decrease of cdc2 expression. Biochem. Biophys. Res. Commun. 2007, 362, 848–852. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.H.; Yu, Y.S.; Jun, H.O.; Kwon, H.J.; Park, K.H.; Kim, K.W. Inhibition of choroidal neovascularization by homoisoflavanone, a new angiogenesis inhibitor. Mol. Vis. 2008, 14, 556–561. [Google Scholar]
- Lee, B.; Basavarajappa, H.D.; Sulaiman, R.S.; Fei, X.; Seo, S.-Y.; Corson, T.W. The first synthesis of the antiangiogenic homoisoflavanone, cremastranone. Org. Biomol. Chem. 2014, 12, 7673–7677. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, H.D.; Sulaiman, R.S.; Qi, X.; Shetty, T.; Sheik Pran Babu, S.; Sishtla, K.L.; Lee, B.; Quigley, J.; Alkhairy, S.; Briggs, C.M.; et al. Ferrochelatase is a therapeutic target for ocular neovascularization. EMBO Mol. Med. 2017, 9, 786–801. [Google Scholar] [CrossRef]
- Sishtla, K.; Lambert-Cheatham, N.; Lee, B.; Han, D.H.; Park, J.; Pasha, S.P.B.S.; Lee, S.; Kwon, S.; Muniyandi, A.; Park, B.; et al. Small-molecule inhibitors of ferrochelatase are antiangiogenic agents. Cell Chem. Biol. 2022, 29, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, H.D.; Lee, B.; Lee, H.; Sulaiman, R.S.; An, H.; Magaña, C.; Shadmand, M.; Vayl, A.; Rajashekhar, G.; Kim, E.-Y.; et al. Synthesis and biological evaluation of novel homoisoflavonoids for retinal neovascularization. J. Med. Chem. 2015, 58, 5015–5027. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Sardar Pasha, S.P.B.; Sishtla, K.L.; Hartman, G.D.; Qi, X.; Boulton, M.E.; Corson, T.W. Decreased expression of soluble epoxide hydrolase suppresses murine choroidal neovascularization. Int. J. Mol. Sci. 2022, 23, 15595. [Google Scholar] [CrossRef]
- Sulaiman, R.S.; Merrigan, S.; Quigley, J.; Qi, X.; Lee, B.; Boulton, M.E.; Kennedy, B.; Seo, S.-Y.; Corson, T.W. A novel small molecule ameliorates ocular neovascularisation and synergises with anti-VEGF therapy. Sci. Rep. 2016, 6, 25509. [Google Scholar] [CrossRef]
- Lee, B.; Sun, W.; Lee, H.; Basavarajappa, H.; Sulaiman, R.S.; Sishtla, K.; Fei, X.; Corson, T.W.; Seo, S.-Y. Design, synthesis and biological evaluation of photoaffinity probes of antiangiogenic homoisoflavonoids. Bioorg. Med. Chem. Lett. 2016, 26, 4277–4281. [Google Scholar] [CrossRef]
- Sulaiman, R.S.; Park, B.; Sheik Pran Babu, S.P.; Si, Y.; Kharwadkar, R.; Mitter, S.K.; Lee, B.; Sun, W.; Qi, X.; Boulton, M.E.; et al. Chemical Proteomics Reveals Soluble Epoxide Hydrolase as a Therapeutic Target for Ocular Neovascularization. ACS Chem. Biol. 2018, 13, 45–52. [Google Scholar] [CrossRef]
- Wagner, K.; Inceoglu, B.; Hammock, B.D. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat. 2011, 96, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.-X.; Song, X.-M.; Wang, L.-C.; Lv, H.-N.; Chen, J.-F.; Liu, D.; Fu, G.; Zhao, M.-B.; Jiang, Y.; Zeng, K.-W.; et al. Highly selective inhibition of IMPDH2 provides the basis of antineuroinflammation therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E5986–E5994. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, I. A total synthesis of (+)-brazilin. J. Org. Chem. 2015, 80, 2001–2005. [Google Scholar] [CrossRef]
- Arredondo, V.; Roa, D.E.; Gutman, E.S.; Huynh, N.O.; Van Vranken, D.L. Total Synthesis of (±)-Brazilin Using [4+ 1] Palladium-Catalyzed Carbenylative Annulation. J. Org. Chem. 2019, 84, 14745–14759. [Google Scholar] [CrossRef]
- Iqbal, M.; Hasanah, N.; Arianto, A.D.; Aryati, A.A.; Puteri, M.U.; Saputri, F.C. Brazilin from Caesalpinia sappan L. as a Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibitor: Pharmacophore-Based Virtual Screening, In silico Molecular Docking, and In Vitro Studies. Adv. Pharmacol. Pharm. Sci. 2023, 2023, 10. [Google Scholar] [CrossRef]
- McMahon, E.; El-Sayed, S.; Green, J.; Hoyle, C.; Fitzpatrick, L.; Jones, E.V.; Corrie, E.; Kelly, R.L.; Chalinor, M.; Freeman, S.; et al. Brazilin is a natural product inhibitor of the NLRP3 inflammasome. Iscience 2024, 27, 108968. [Google Scholar] [CrossRef]
- Cui, Z.; Guo, F.Y.; Li, L.; Jin, C.H.; Wang, X.; Liu, F. Brazilin-7-acetate, a novel potential drug of Parkinson’s disease, hinders the formation of α-synuclein fibril, mitigates cytotoxicity, and decreases oxidative stress. Eur. J. Med. Chem. 2024, 264, 115965. [Google Scholar] [CrossRef]
- Guo, Q.; Zhang, Y.-C.; Wang, W.; Wang, Y.-Q.; Liu, Y.; Yang, Z.; Zhao, M.-M.; Feng, N.; Wang, Y.-H.; Zhang, X.-W.; et al. Deoxyhypusine hydroxylase as a novel pharmacological target for ischemic stroke via inducing a unique post-translational hypusination modification. Pharmacol. Res. 2022, 176, 106046. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Peng, X.; Guo, Y.; Gong, S.; Cao, S.; Qiu, F. Currently available strategies for target identification of bioactive natural products. Front. Chem. 2021, 9, 761609. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe Structure | Methods | Target | Disease |
|---|---|---|---|
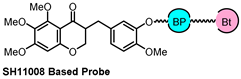 | PAL and X-ray co-crystal | FECH | angiogenesis |
 | PAL and MD simulation | sEH | angiogenesis |
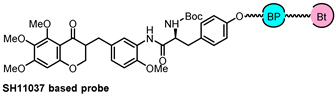
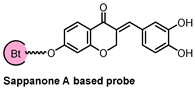 | CETSA, DARTS, and MD simulation | IMPDH2 | inflammatory |
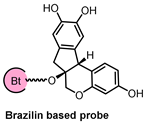 | Microarray, CETSA, Molecular Docking Simulation | DOHH | Ischemic stroke |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fei, X.; Kwon, S.; Jang, J.; Seo, M.; Yu, S.; Corson, T.W.; Seo, S.-Y. Exploring the Antiangiogenic and Anti-Inflammatory Potential of Homoisoflavonoids: Target Identification Using Biotin Probes. Biomolecules 2024, 14, 785. https://doi.org/10.3390/biom14070785
Fei X, Kwon S, Jang J, Seo M, Yu S, Corson TW, Seo S-Y. Exploring the Antiangiogenic and Anti-Inflammatory Potential of Homoisoflavonoids: Target Identification Using Biotin Probes. Biomolecules. 2024; 14(7):785. https://doi.org/10.3390/biom14070785
Chicago/Turabian StyleFei, Xiang, Sangil Kwon, Jinyoung Jang, Minyoung Seo, Seongwon Yu, Timothy W. Corson, and Seung-Yong Seo. 2024. "Exploring the Antiangiogenic and Anti-Inflammatory Potential of Homoisoflavonoids: Target Identification Using Biotin Probes" Biomolecules 14, no. 7: 785. https://doi.org/10.3390/biom14070785
APA StyleFei, X., Kwon, S., Jang, J., Seo, M., Yu, S., Corson, T. W., & Seo, S.-Y. (2024). Exploring the Antiangiogenic and Anti-Inflammatory Potential of Homoisoflavonoids: Target Identification Using Biotin Probes. Biomolecules, 14(7), 785. https://doi.org/10.3390/biom14070785