
Phylodynamics and Coat Protein Analysis of Babaco Mosaic Virus in Ecuador

 , , , ,
, , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Nucleotide Sequences and Recombination Analyses
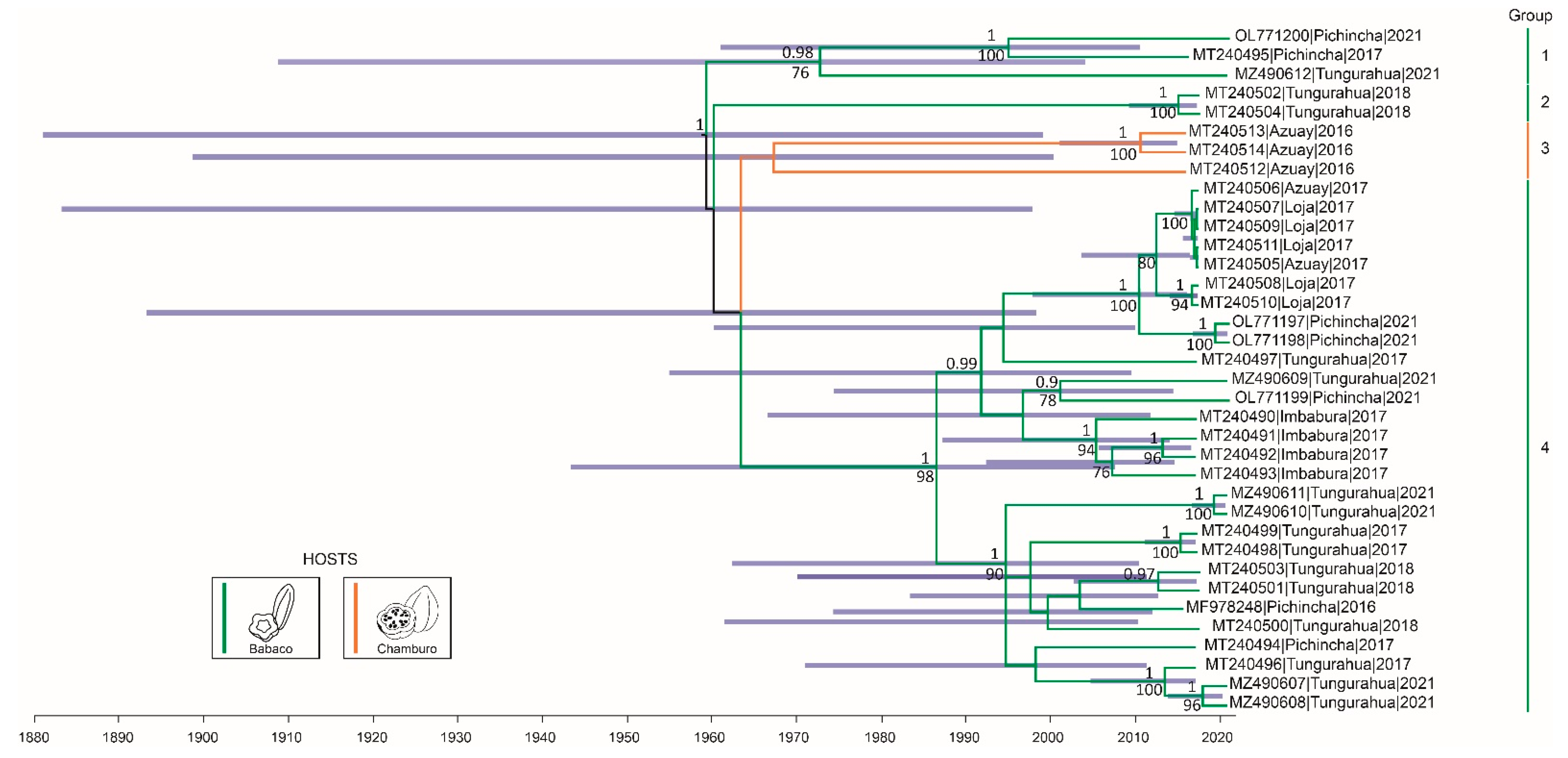
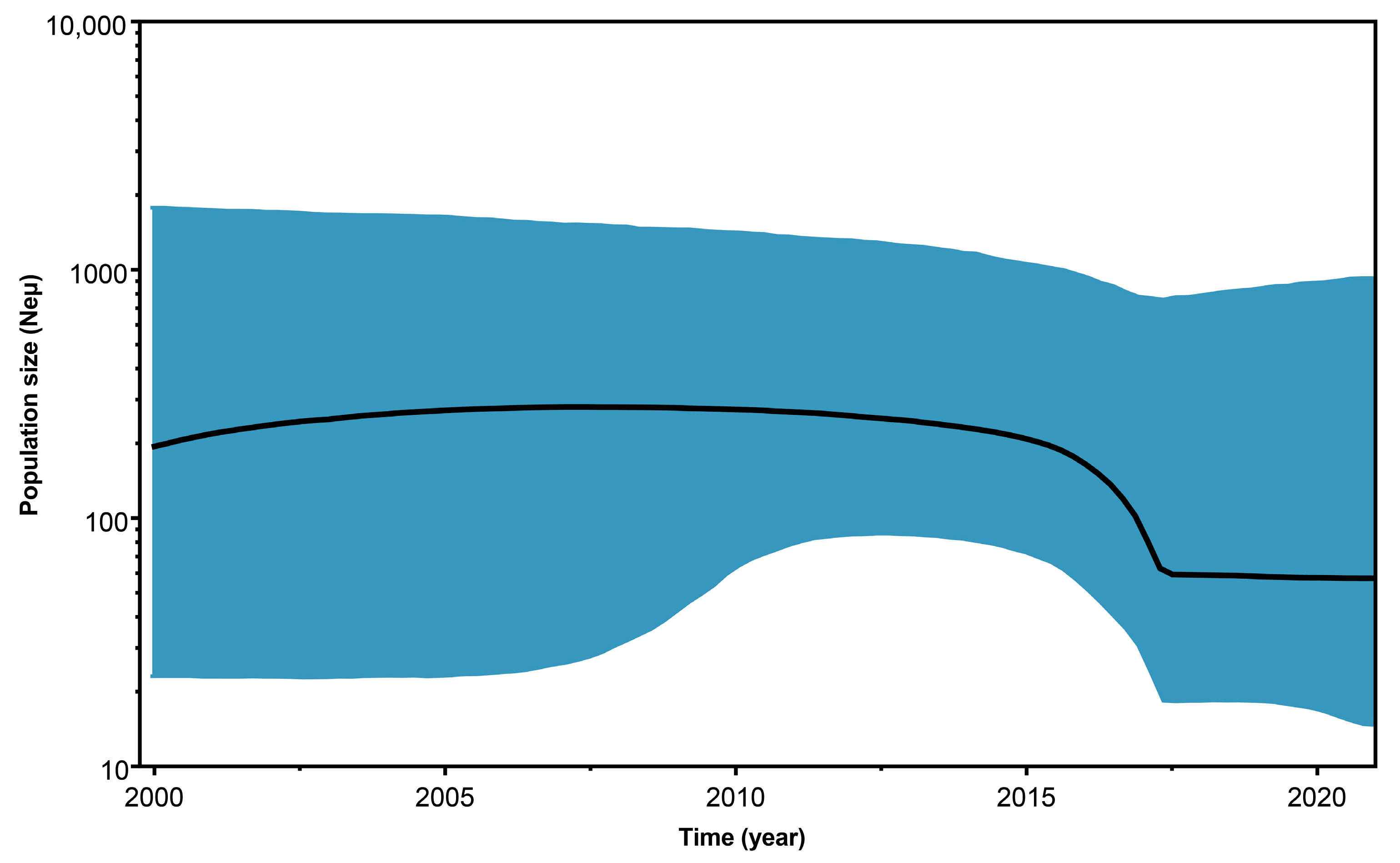
2.2. Phylogenetic Analysis and Geographical Spread of BabMV
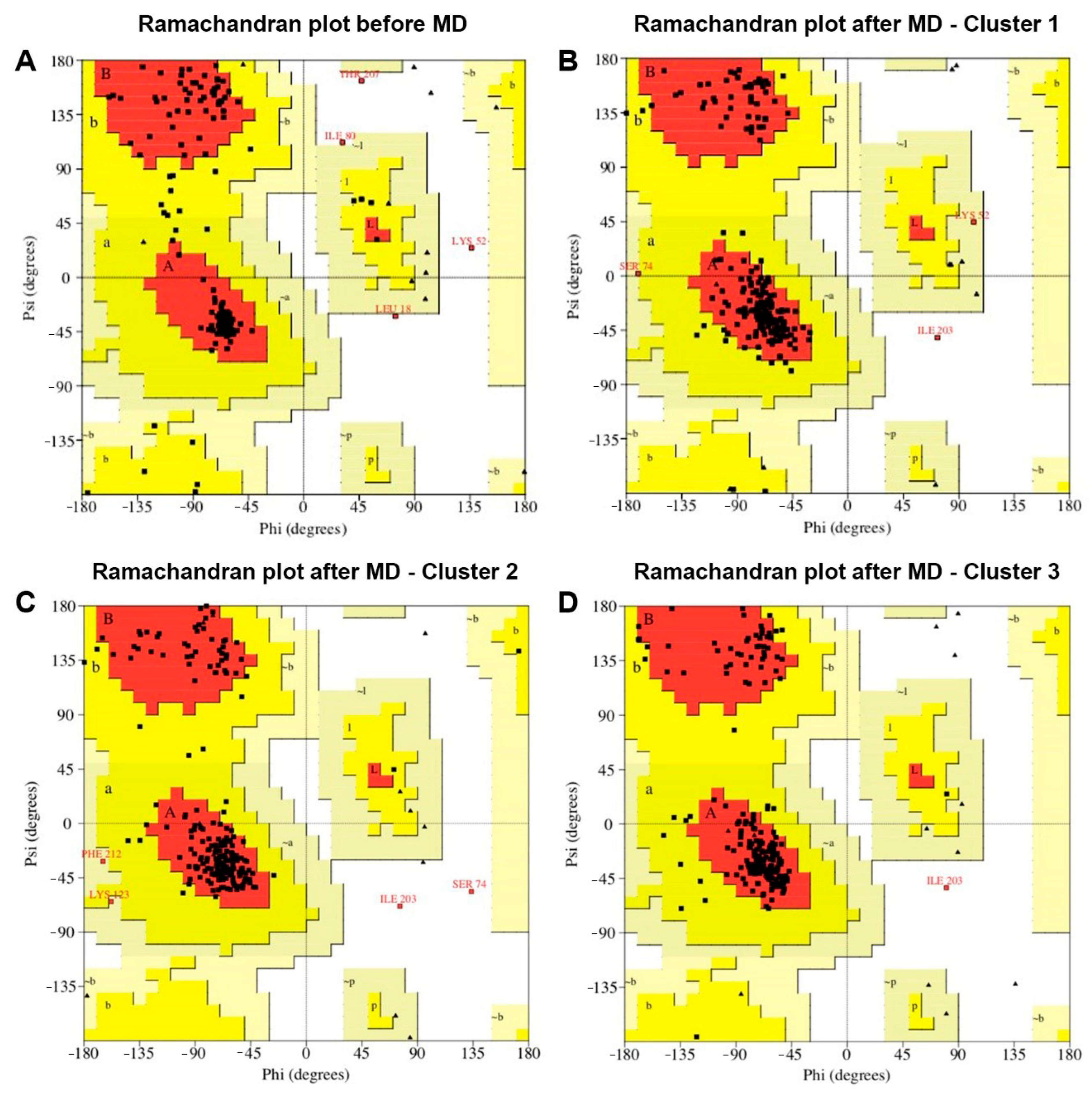
2.3. Three-Dimensional Structure of the BabMV Coat Protein
3. Discussion
4. Materials and Methods
4.1. Collection of Virus Isolates, Viral RNA, and Sequencing
4.2. Recombination Analysis
4.3. Phylogenetic Analysis
4.4. Molecular Modeling of the Babaco Mosaic Virus Coat Protein
4.5. Molecular Dynamics Simulation
4.6. Three-Dimensional Structural Alignment and Antibody Epitope Prediction
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bacon, J. Exotic Fruits and Vegetables A-Z; UPSO Ltd.: St Leonards on Sea, UK, 2004. [Google Scholar]
- Carvalho, F.A.; Renner, S.S. A dated phylogeny of the papaya family (Caricaceae) reveals the crop’s closest relatives and the family’s biogeographic history. Mol. Phylogenet. Evol. 2012, 65, 46–53. [Google Scholar] [CrossRef]
- Villareal, L.; Dhuique-Mayer, C.; Dornier, M.; Ruales, J.; Reynes, M. Évaluation de l’intérêt du babaco (Carica pentagona Heilb.). Fruits 2003, 58, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Vásquez, W.; Villavicencio, A. Guía de Cultivos; INIAP: Quito, Ecuador, 2008. [Google Scholar]
- Kempler, C.; Kabaluk, J.T.; Nelson, M. Greenhouse cultivation of babaco (Carica × heilbornii Badillo n.m. pentagona (Heilborn)): Effect of media, container size, stem number, and plant density. N. Z. J. Crop Hort. 1993, 21, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Soria, N. Babaco, fruto con potencial en el Ecuador y el mundo. Rev. Inf. INIAP 1997, 9, 35–43. [Google Scholar]
- National Research Council. Highland Papayas; National Research Council: Washington, DC, USA, 1989; pp. 252–261. [Google Scholar]
- Dhuique-Mayer, C.; Caro, Y.; Pina, M.; Ruales, J.; Dornier, M.; Graille, J. Biocatalytic properties of lipase in crude latex from babaco fruit (Carica pentagona). Biotechnol. Lett. 2001, 23, 1021–1024. [Google Scholar] [CrossRef]
- Alvarez-Quinto, R.A.; Cornejo-Franco, J.F.; Quito-Avila, D.F. Characterization of a not so new potexvirus from babaco (Vasconcellea × heilbornii). PLoS ONE 2017, 12, e0189519. [Google Scholar] [CrossRef] [Green Version]
- Cornejo-Franco, J.F.; Medina-Salguero, A.; Flores, F.; Chica, E.; Grinstead, S.; Mollov, D.; Quio-Avila, D.F. Exploring the virome of Vasconcellea × heilbornii: The first step towards a sustainable production program for babaco in Ecuador. Eur. J. Plant Pathol. 2020, 157, 961–968. [Google Scholar] [CrossRef]
- Marina, B.; Giordano, P. An elongated virus associated with yellow mosaic of babaco. Acta Hortic. 1989, 235, 149–156. [Google Scholar] [CrossRef]
- Pisi, A.; Vicchi, V. Virus-like particles found in babaco plants (Carica pentagona H.) in Italy. Adv. Hort. Sci. 1989, 3, 73–77. [Google Scholar] [CrossRef]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Lemey, P. Towards a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Sargsyan, K.; Cédric, G.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Agirrezabala, X.; Méndez-López, E.; Lasso, G.; Sánchez-Pina, M.A.; Aranda, M.; Valle, M. The near-atomic cryoEM structure of a flexible filamentous plant virus shows homology of its coat protein with nucleoproteins of animal viruses. eLife 2015, 4, e11795. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, F.; Chen, C.C.; Yu, X.; Frenz, B.; Hsu, Y.H.; Lin, N.S.; Egelman, E.H. The molecular basis for flexibility in the flexible filamentous plant viruses. Nat. Struct. Mol. Biol. 2015, 22, 642–644. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, T.; Bohon, J.; Gagné, M.È.; Bolduc, M.; Leclerc, D.; Li, H. Crystal structure of the coat protein of the flexible filamentous papaya mosaic virus. J. Mol. Biol. 2012, 422, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Zamora, M.; Méndez-López, E.; Agirrezabala, X.; Cuesta, R.; Lavín, J.L.; Sánchez-Pina, M.A.; Aranda, M.A.; Valle, M. Potyvirus virion structure shows conserved protein fold and RNA binding site in ssRNA viruses. Sci. Adv. 2017, 3, eaao2182. [Google Scholar] [CrossRef] [Green Version]
- Bancroft, J.B.; Rouleau, M.; Johnston, R.; Prins, L.; Mackie, G.A. The entire nucleotide sequence of foxtail mosaic virus RNA. J. Gen. Virol. 1991, 72, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Chia, T.F.; Chan, Y.S.; Chua, N.H. Characterization of cymbidium mosaic virus coat protein gene and its expression in transgenic tobacco plants. Plant. Mol. Biol. 1992, 18, 1091–1909. [Google Scholar] [CrossRef]
- Gómez, P.; Sempere, R.N.; Aranda, M.A.; Elena, S.F. Phylodynamics of Pepino mosaic virus in Spain. Eur. J. Plant Pathol. 2012, 134, 445–449. [Google Scholar] [CrossRef]
- He, M.; He, C.; Ding, N. Evolution of Potato virus X. Mol. Phylogenet. Evol. 2022, 167, 107336. [Google Scholar] [CrossRef]
- Minicka, J.; Hasiów-Jaroszewska, B.; Borodynko-Filas, N.; Pospieszny, H.; Hanssen, I. Rapid evolutionary dynamics of the Pepino mosaic virus—Status and future perspectives. J. Plant Prot. Res. 2016, 56, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Simmons, H.E.; Holmes, E.C.; Stephenson, A.G. Rapid evolutionary dynamics of zucchini yellow mosaic virus. J. Gen. Virol. 2008, 89, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Pagán, I.; Firth, C.; Holmes, E.C. Phylogenetic Analysis Reveals Rapid Evolutionary Dynamics in the Plant RNA Virus Genus Tobamovirus. J. Mol. Evol. 2010, 71, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Fargette, D.; Pinel, A.; Rakotomalala, M.; Sangu, E.; Traoré, O.; Sérémé, D.; Sorho, F.; Issaka, S.; Hébrard, E.; Séré, Y.; et al. Rice yellow mottle virus, an RNA plant virus, evolves as rapidly as most RNA animal viruses. J. Virol. 2008, 82, 3584–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Blanchard-Letort, A.; Liu, Y.; Zhou, G.; Wang, X.; Elena, S.F. Dynamics of Molecular Evolution and Phylogeography of Barley yellow dwarf virus-PAV. PLoS ONE 2011, 6, e16896. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Holmes, E.C. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J. Virol. 2008, 82, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Holmes, E.C. Validation of high rates of nucleotide substitution in geminiviruses: Phylogenetic evidence from East African cassava mosaic viruses. J. Gen. Virol. 2009, 90, 1539–1547. [Google Scholar] [CrossRef]
- Almeida, R.P.P.; Bennett, G.M.; Anhalt, M.D.; Tsai, C.W.; O’Grady, P. Spread of an introduced vector-borne banana virus in Hawaii. Mol. Ecol. 2009, 18, 136–146. [Google Scholar] [CrossRef]
- Grigoras, I.; Timchenko, T.; Grande-Pérez, A.; Katul, L.; Vetten, H.J.; Gronenborn, B. High variability and rapid evolution of a nanovirus. J. Virol. 2010, 84, 9105–9117. [Google Scholar] [CrossRef] [Green Version]
- Tobar, M. Análisis de Competitividad de los Productores de Babaco de San Pablo de Tenta (Saraguro, Loja) con Enfoque de Agrocadena. Ph.D. Thesis, Universidad Andina Simón Bolívar, Sucre, Bolivia, 2008. [Google Scholar]
- López, H.; Viera, D.; Bassante, V.; Ortiz, M. Research to determine the feasibility of the establishment of a microenterprise of a babaco jam in the Pillaro canton. Rev. Cienc. Tecnol 2018, 20, 51–63. [Google Scholar] [CrossRef]
- Ampuero, P. Proyecto de Elaboración de Mermelada de Babaco como Producto no Tradicional de Exportación al Mercado Europeo. Bachelor’s Thesis, Escuela Superior Politécnica del Litoral, Guayaquil, Ecuador, 2004. [Google Scholar]
- Medina, L.; Villafuerte, R. Estudio de Factibilidad para la Instalación de una Planta Procesadora de Frutas para Producir Babaco en Almíbar. Bachelor’s Thesis, Universidad San Francisco de Quito, Quito, Ecuador, 2005. [Google Scholar]
- Aguilera-Pesantes, D.; Méndez, M.A. Structure and sequence based functional annotation of Zika virus NS2b protein: Computational insights. Biochem. Biophys. Res. Commun. 2017, 492, 659–667. [Google Scholar] [CrossRef]
- Mishra, P.M.; Uversky, V.N.; Giri, R. Molecular recognition features in Zika virus proteome. J. Mol. Biol. 2018, 430, 2372–2388. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible Emergence of New Geminiviruses by Frequent Recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Nat. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Oña-Chuquimarca, S.; Ayala-Ruano, S.; Goossens, J.; Pauwels, L.; Goossens, A.; Leon-Reyes, A.; Méndez, M.A. The molecular basis of JAZ-MYC coupling, a protein-protein interface essential for plant response to stressors. Front. Plant Sci. 2020, 11, 1139. [Google Scholar] [CrossRef]
- Bhutani, I.; Loharch, S.; Gupta, P.; Madathil, R.; Parkesh, R. Structure, dynamics, and interaction of Mycobacterium tuberculosis (Mtb) DprE1 and DprE2 examined by molecular modeling, simulation, and electrostatic studies. PLoS ONE 2015, 10, e0119771. [Google Scholar] [CrossRef]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS—A message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Huber, T.; Torda, A.E.; van Gunsteren, W.F.J. Local elevation: A method for improving the searching properties of molecular dynamics simulation. J. Comput. Aided Mol. Des. 1994, 8, 695–708. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, J.; Bui, H.H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clock Model | Demographic Model | Path Sampling | 2lnBF | Stepping Stone | 2lnBF |
|---|---|---|---|---|---|
| Strict | Constant size | −3336.8 | 63.8 | −3336.8 | 63.4 |
| Strict | Exponential growth | −3336.7 | 63.8 | −3336.7 | 63.2 |
| Strict | Bayesian skyline | −3304.9 | N/A | −3305.1 | N/A |
| Protein | Min RMSF (Å) | Max RMSF (Å) | Average RMSF (Å) (n = 64 Clusters) |
|---|---|---|---|
| CP | 0.634 | 6.328 | 2.316 |
| RDRP | 0.114 | 1.195 | 0.327 |
| TGB1 | 0.505 | 5.536 | 2.079 |
| ALKb-domain | 0.721 | 3.692 | 1.777 |
| Isolate | Origin | Latitude–Longitude | Collection Date | Host | Accession Number | Reference |
|---|---|---|---|---|---|---|
| Tandapi | Pichincha | 0.414 S 78.799 W | 1 March 2016 | Babaco | MF978248 | Alvarez-Quinto et al. (2017) |
| IB1-7 | Imbabura | 0.357 N 78.241 W | 12 August 2017 | Babaco | MT240490 | This study |
| IB2-7 | Imbabura | 0.372 N 78.241 W | 12 August 2017 | Babaco | MT240491 | This study |
| IB3-7 | Imbabura | 0.359 N 78.217 W | 12 August 2017 | Babaco | MT240492 | This study |
| IB4-7 | Imbabura | 0.334 N 78.239 W | 12 August 2017 | Babaco | MT240493 | This study |
| PB1-7 | Pichincha | 0.070 S 78.572 W | 12 August 2017 | Babaco | MT240494 | This study |
| PB2-7 | Pichincha | 0.216 S 78.412 W | 9 March 2017 | Babaco | MT240495 | This study |
| PB3-1 | Pichincha | 0.038 S 78.564 W | 15 June 2021 | Babaco | OL771197 | This study |
| PB4-1 | Pichincha | 0.038 S 78.564 W | 15 June 2021 | Babaco | OL771198 | This study |
| PB5-1 | Pichincha | 0.038 S 78.564 W | 15 June 2021 | Babaco | OL771199 | This study |
| PB6-1 | Pichincha | 0.038 S 78.564 W | 15 June 2021 | Babaco | OL771200 | This study |
| TB1-7 | Tungurahua | 1.176 S 78.559 W | 17 September 2017 | Babaco | MT240496 | This study |
| TB2-7 | Tungurahua | 1.159 S 78.549 W | 17 September 2017 | Babaco | MT240497 | This study |
| TB3-7 | Tungurahua | 1.294 S 78.488 W | 17 September 2017 | Babaco | MT240498 | This study |
| TB4-7 | Tungurahua | 1.295 S 78.487 W | 17 September 2017 | Babaco | MT240499 | This study |
| TB1-8 | Tungurahua | 1.418 S 78.405 W | 24 January 2018 | Babaco | MT240500 | This study |
| TB2-8 | Tungurahua | 1.414 S 78.429 W | 24 January 2018 | Babaco | MT240501 | This study |
| TB3-8 | Tungurahua | 1.404 S 78.406 W | 24 January 2018 | Babaco | MT240502 | This study |
| TB4-8 | Tungurahua | 1.404 S 78.399 W | 24 January 2018 | Babaco | MT240503 | This study |
| TB5-8 | Tungurahua | 1.399 S 78.406 W | 24 January 2018 | Babaco | MT240504 | This study |
| TB1-1 | Tungurahua | 1.180 S 78.560 W | 13 April 2021 | Babaco | MZ490607 | This study |
| TB2-1 | Tungurahua | 1.180 S 78.560 W | 13 April 2021 | Babaco | MZ490608 | This study |
| TB3-1 | Tungurahua | 1.320 S 78.470 W | 13 April 2021 | Babaco | MZ490609 | This study |
| TB4-1 | Tungurahua | 1.340 S 78.470 W | 13 April 2021 | Babaco | MZ490610 | This study |
| TB5-1 | Tungurahua | 1.330 S 78.470 W | 13 April 2021 | Babaco | MZ490611 | This study |
| TB6-1 | Tungurahua | 1.340 S 78.470 W | 13 April 2021 | Babaco | MZ490612 | This study |
| AB1-7 | Azuay | 2.727 S 78.779 W | 20 November 2017 | Babaco | MT240505 | This study |
| AB2-7 | Azuay | 2.748 S 78.773 W | 20 November 2017 | Babaco | MT240506 | This study |
| LB1-7 | Loja | 3.614 S 79.261 W | 20 November 2017 | Babaco | MT240507 | This study |
| LB2-7 | Loja | 3.610 S 79.265 W | 20 November 2017 | Babaco | MT240508 | This study |
| LB3-7 | Loja | 3.598 S 79.312 W | 20 November 2017 | Babaco | MT240509 | This study |
| LB4-7 | Loja | 3.594 S 79.274 W | 20 November 2017 | Babaco | MT240510 | This study |
| LB5-7 | Loja | 3.584 S 79.278 W | 20 November 2017 | Babaco | MT240511 | This study |
| AC1-6 | Azuay | 2.796 S 78.768 W | 15 June 2016 | Chamburo | MT240512 | This study |
| AC2-6 | Azuay | 2.796 S 78.768 W | 15 June 2016 | Chamburo | MT240513 | This study |
| AC3-6 | Azuay | 2.796 S 78.768 W | 15 June 2016 | Chamburo | MT240514 | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosquera-Yuqui, F.; Flores, F.J.; Moncayo, E.A.; Garzón-Proaño, B.A.; Méndez, M.A.; Guevara, F.E.; Quito-Avila, D.F.; Viera, W.; Cornejo-Franco, J.F.; Izquierdo, A.R.; et al. Phylodynamics and Coat Protein Analysis of Babaco Mosaic Virus in Ecuador. Plants 2022, 11, 1646. https://doi.org/10.3390/plants11131646
Mosquera-Yuqui F, Flores FJ, Moncayo EA, Garzón-Proaño BA, Méndez MA, Guevara FE, Quito-Avila DF, Viera W, Cornejo-Franco JF, Izquierdo AR, et al. Phylodynamics and Coat Protein Analysis of Babaco Mosaic Virus in Ecuador. Plants. 2022; 11(13):1646. https://doi.org/10.3390/plants11131646
Chicago/Turabian StyleMosquera-Yuqui, Francisco, Francisco J. Flores, Eduardo A. Moncayo, Brighitte A. Garzón-Proaño, Miguel A. Méndez, Fiama E. Guevara, Diego F. Quito-Avila, William Viera, Juan F. Cornejo-Franco, Andrés R. Izquierdo, and et al. 2022. "Phylodynamics and Coat Protein Analysis of Babaco Mosaic Virus in Ecuador" Plants 11, no. 13: 1646. https://doi.org/10.3390/plants11131646