

Genome-Wide Identification and Molecular Evolutionary History of the Whirly Family Genes in Brassica napus
 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Genome-Wide Identification and Whirly Gene Family Analysis
2.2. Protein Physicochemical Properties and Gene Structures Analysis
2.3. Analysis of the Chromosomal Localization and Phylogeny of the Whirly Gene Family
2.4. Protein 3D Structure Prediction and Cis-Acting Element Analysis
2.5. Collinearity Analysis and Protein Interaction Network Prediction of Whirly Gene Family
2.6. Subcellular Localization Analysis
2.7. Expression Patterns and qRT-PCR Analysis of Whirly Gene Family in B. napus
3. Results
3.1. Identification and Physicochemical Characterization of Whirly Genes
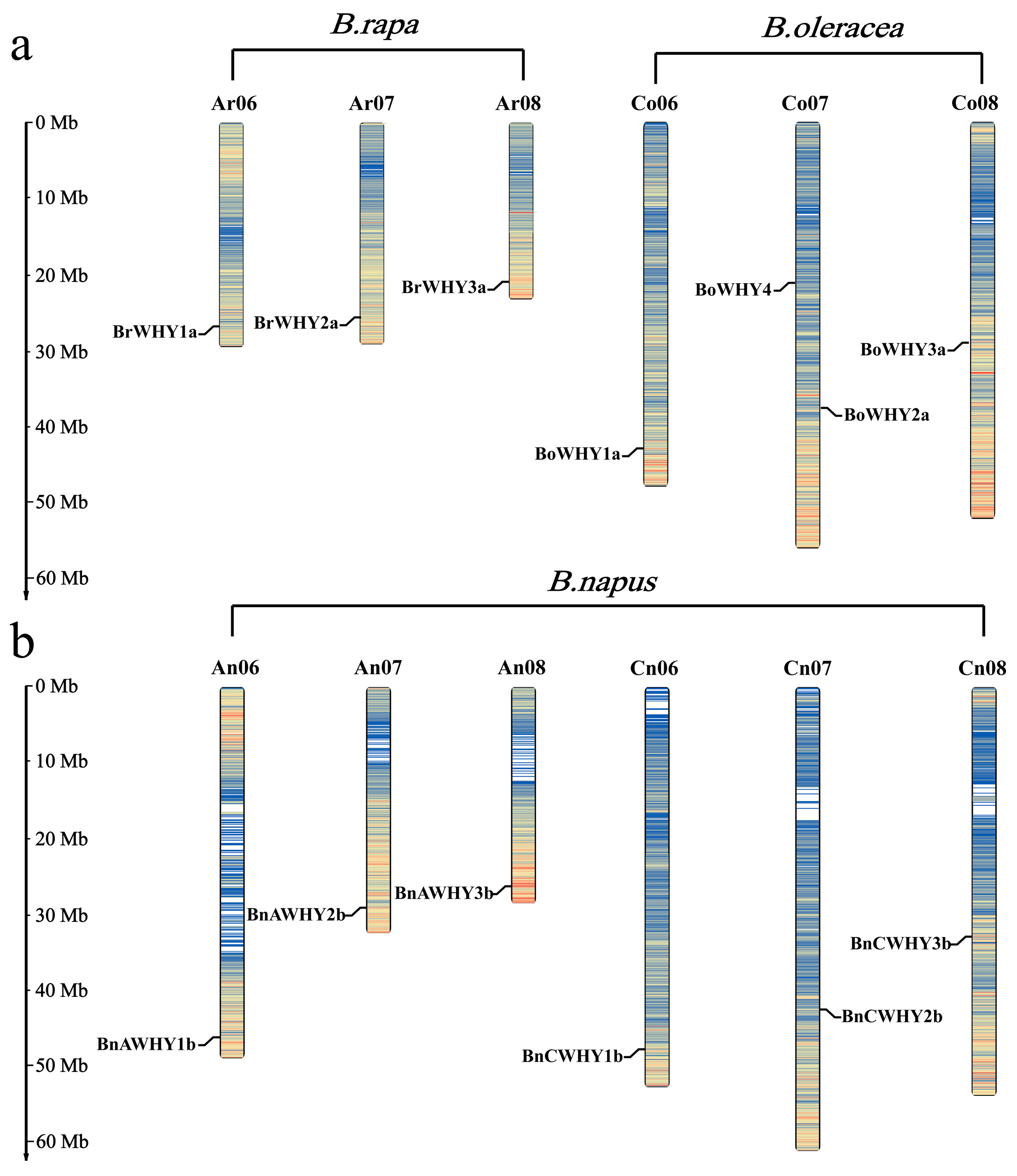
3.2. Chromosomal Localization of the Whirly Genes
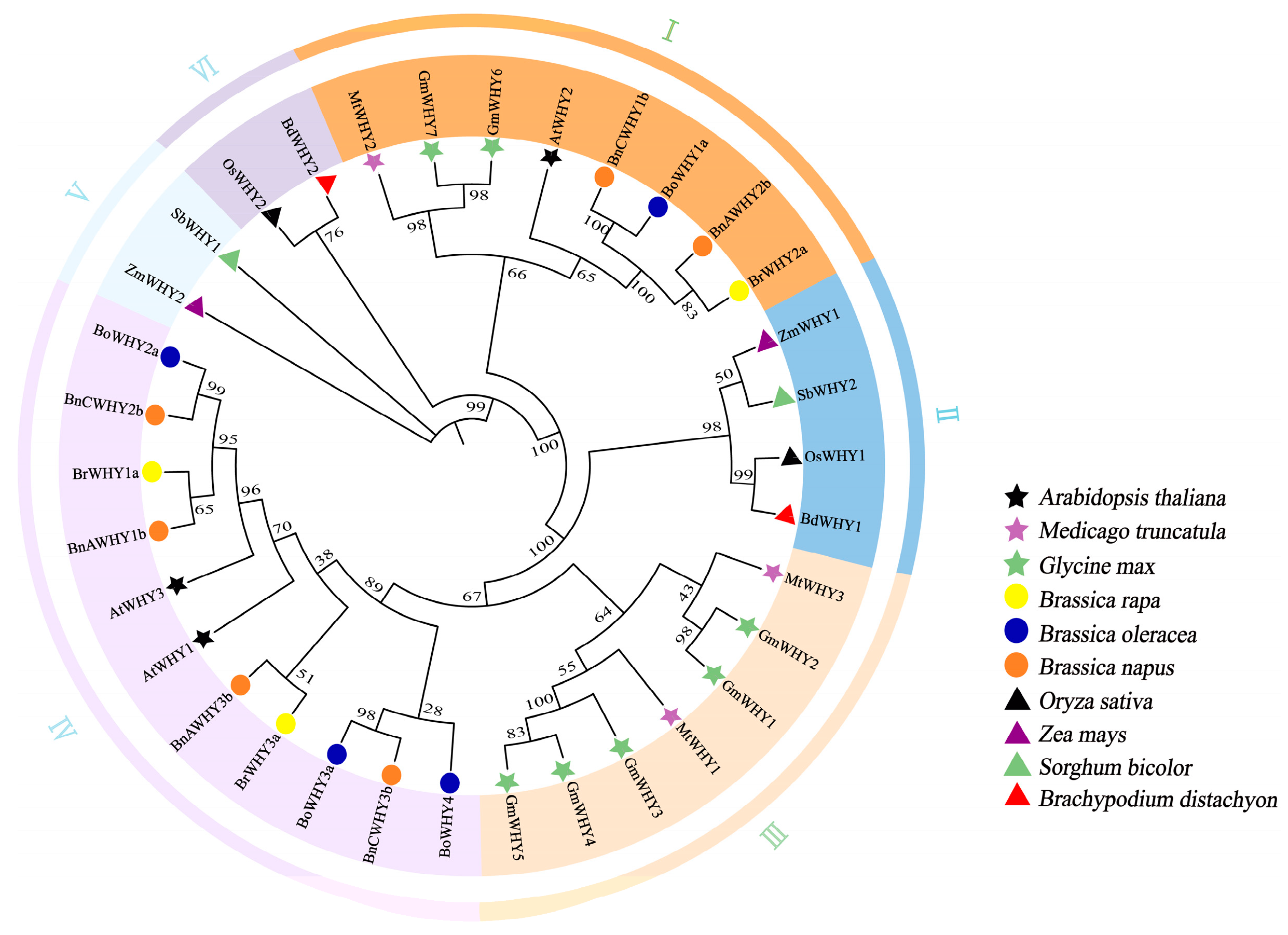
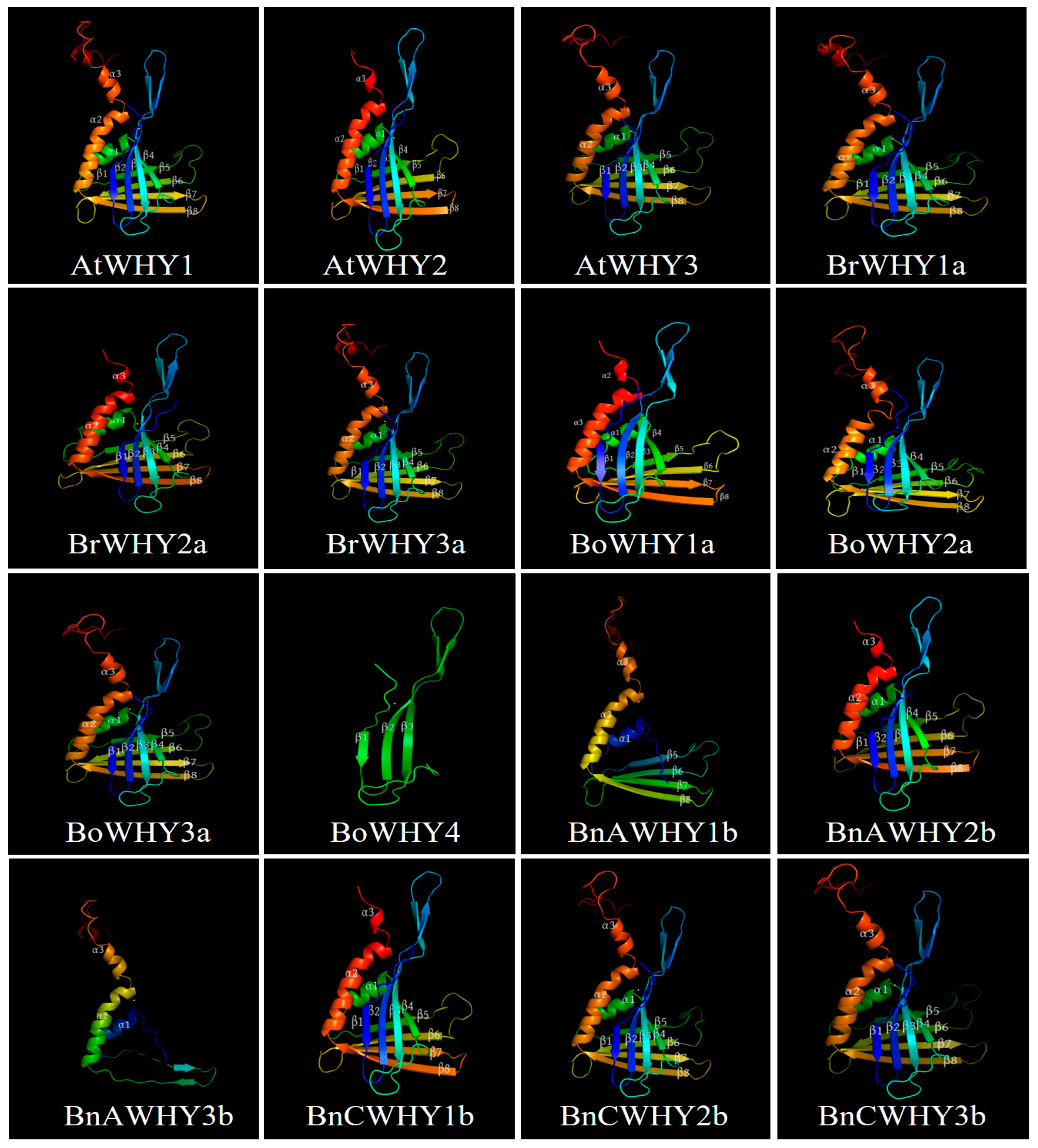
3.3. Phylogenetic and Homology Modeling Analysis of Whirly Proteins
3.4. Motif Composition, Conserved Structural Domains, and Gene Structures of the Whirly Gene Family
3.5. Cis-Acting Elements in the Promoter Region of the Whirly Gene Family
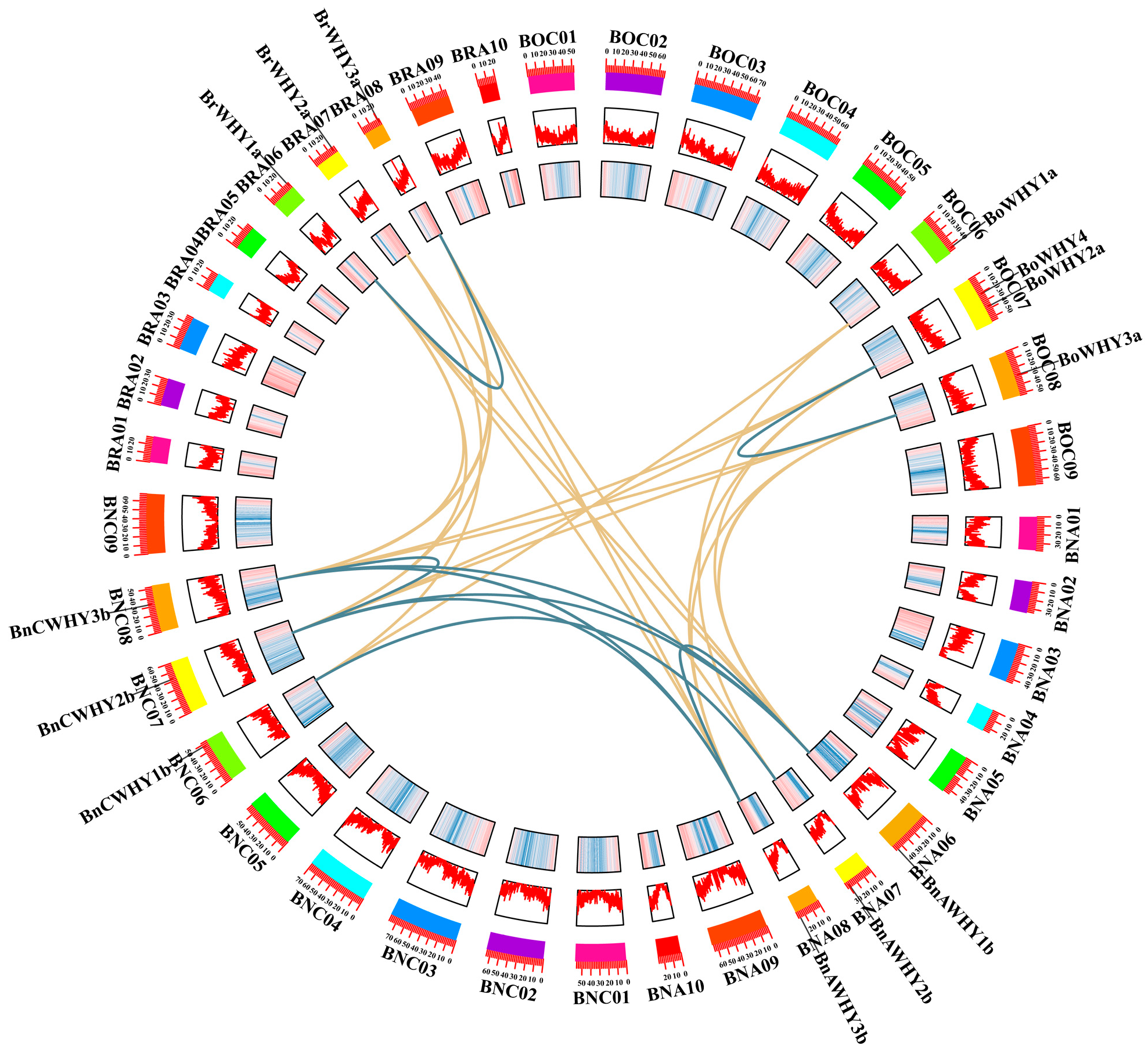
3.6. Collinearity Relationship of the Whirly Gene Family
3.7. Prediction of Whirly Protein Interactions Network in B. napus
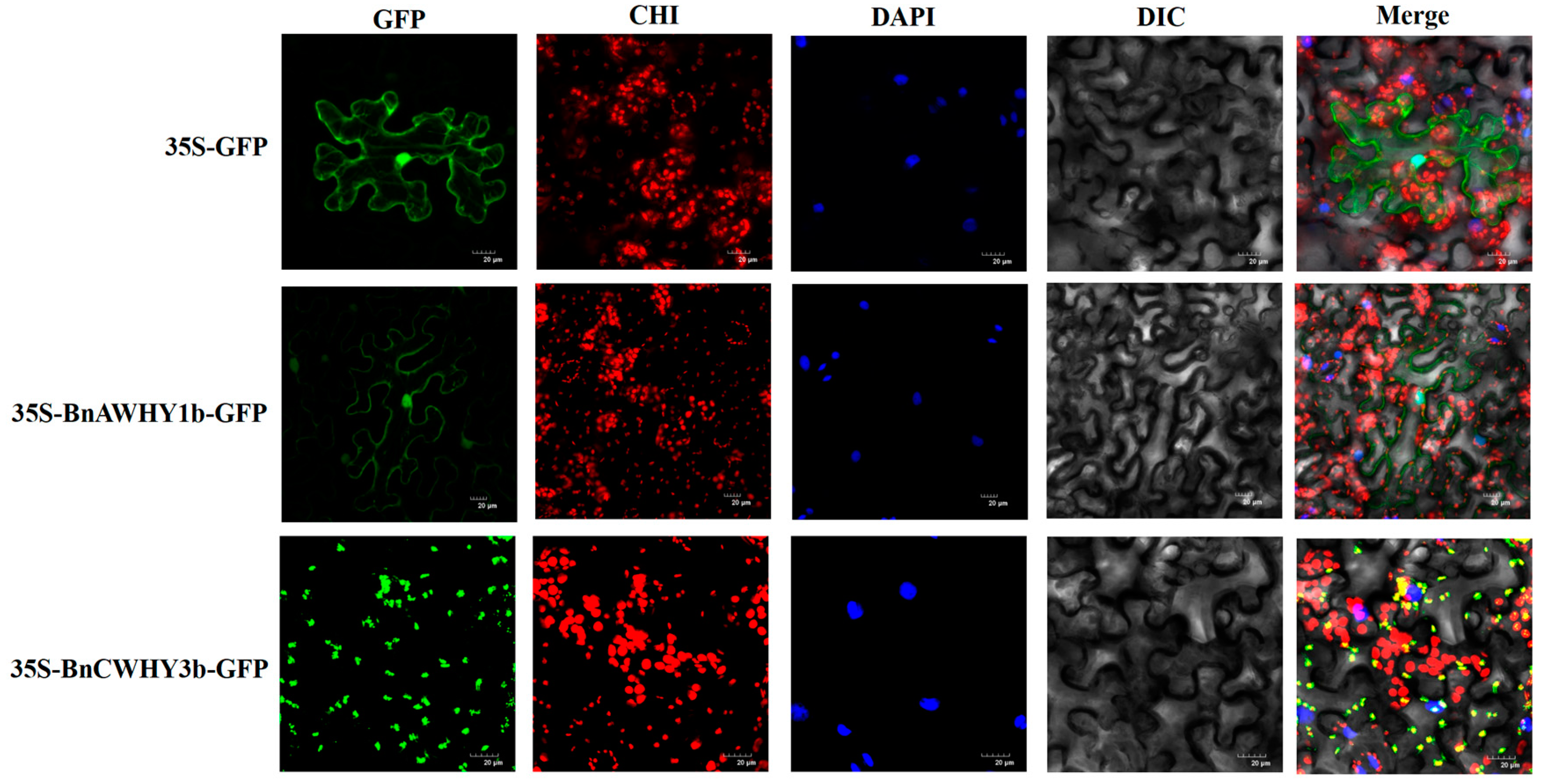
3.8. Subcellular Localization of BnAWHY1b and BnCWHY3b
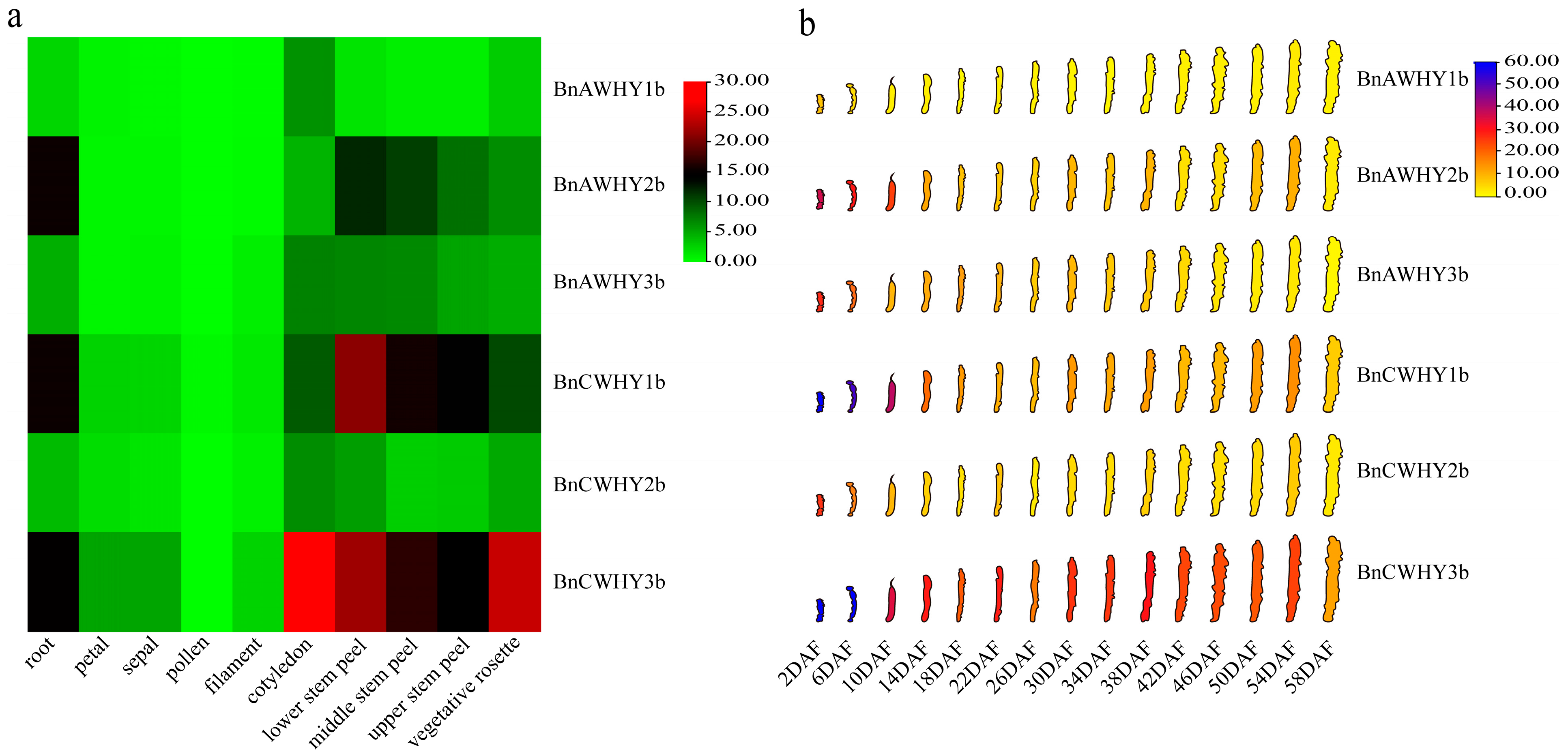
3.9. Tissue Expression Patterns of the Whirly Gene Family in B. napus
3.10. Expression Pattern Analysis of BnWHY Genes by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mayrose, I.; Barker, M.S.; Otto, S.P. Probabilistic models of chromosome number evolution and the inference of polyploidy. Syst. Biol. 2010, 59, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Otto, S.P.; Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Gong, Q.; Qin, W.; Yang, Z.; Cheng, Y.; Lu, L.; Ge, X.; Zhang, C.; Wu, Z.; Li, F. Genome-wide analysis of WOX genes in upland cotton and their expression pattern under different stresses. BMC Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Niu, Y.; Zheng, Y. Multiple functions of myb transcription factors in abiotic stress responses. Int. J. Mol. Sci. 2021, 22, 6125. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, T.; Kan, J.; Yao, Y.; Guo, D.; Yang, Y.; Ling, X.; Wang, J.; Zhang, B. The GhMYB36 transcription factor confers resistance to biotic and abiotic stress by enhancing PR1 gene expression in plants. Plant Biotechnol. J. 2022, 20, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Dudhate, A.; Shinde, H.; Yu, P.; Tsugama, D.; Gupta, S.K.; Liu, S.; Takano, T. Comprehensive analysis of NAC transcription factor family uncovers drought and salinity stress response in pearl millet (Pennisetum glaucum). BMC Genom. 2021, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, Y.; Zou, W.; Pan, Y.B.; Lin, P.; Xu, L.; Grisham, M.P.; Ding, Q.; Su, Y.; Que, Y. Genome-wide characterization of sugarcane catalase gene family identifies a ScCAT1 gene associated disease resistance. Int. J. Biol. Macromol. 2023, 232, 123398. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.; Kilbienski, I.; Mulisch, M.; Rödiger, A.; Schäfer, A.; Krupinska, K. DNA-binding proteins of the Whirly family in Arabidopsis thaliana are targeted to the organelles. FEBS Lett. 2005, 579, 3707–3712. [Google Scholar] [CrossRef]
- Yoo, H.H.; Kwon, C.; Lee, M.M.; Chung, I.K. Single-stranded DNA binding factor AtWHY1 modulates telomere length homeostasis in Arabidopsis. Plant J. 2007, 49, 442–451. [Google Scholar] [CrossRef]
- Ren, Y.; Li, Y.; Jiang, Y.; Wu, B.; Miao, Y. Phosphorylation of WHIRLY1 by CIPK14 shifts its localization and dual functions in Arabidopsis. Mol. Plant 2017, 10, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Desveaux, D.; Maréchal, A.; Brisson, N. Whirly transcription factors: Defense gene regulation and beyond. Trends Plant Sci. 2005, 10, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Jiang, J.; Ren, Y.; Zhao, Z. The single-stranded DNA-binding protein WHIRLY1 represses WRKY53 expression and delays leaf senescence in a developmental stage-dependent manner in Arabidopsis. Plant Physiol. 2013, 163, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Janack, B.; Sosoi, P.; Krupinska, K.; Humbeck, K. Knockdown of WHIRLY1 affects drought stress-induced leaf senescence and histone modifications of the senescence-associated gene HvS40. Plants 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Hou, M.M.; Tan, B.C. The requirement of WHIRLY1 for embryogenesis is dependent on genetic background in maize. PLoS ONE 2013, 8, e67369. [Google Scholar] [CrossRef] [PubMed]
- Isemer, R.; Krause, K.; Grabe, N.; Kitahata, N.; Asami, T.; Krupinska, K. Plastid located WHIRLY1 enhances the responsiveness of Arabidopsis seedlings toward abscisic acid. Front. Plant Sci. 2012, 3, 283. [Google Scholar] [CrossRef]
- Prikryl, J.; Watkins, K.P.; Friso, G.; van Wijk, K.J.; Barkan, A. A member of the Whirly family is a multifunctional RNA- and DNA-binding protein that is essential for chloroplast biogenesis. Nucleic Acids Res. 2008, 36, 5152–5165. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Guo, L.; Shen, Z.R.; Wang, D.Y.; Zhang, Q.; Sodmergen. Elevation of pollen mitochondrial DNA copy number by WHIRLY2: Altered respiration and pollen tube growth in Arabidopsis. Plant Physiol. 2015, 169, 660–673. [Google Scholar] [CrossRef]
- Maréchal, A.; Parent, J.S.; Véronneau-Lafortune, F.; Joyeux, A.; Lang, B.F.; Brisson, N. Whirly proteins maintain plastid genome stability in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 14693–14698. [Google Scholar] [CrossRef]
- Maréchal, A.; Parent, J.S.; Sabar, M.; Véronneau-Lafortune, F.; Abou-Rached, C.; Brisson, N. Overexpression of mtDNA-associated AtWhy2 compromises mitochondrial function. BMC Plant Biol. 2008, 8, 42. [Google Scholar] [CrossRef]
- Cappadocia, L.; Maréchal, A.; Parent, J.S.; Lepage, E.; Sygusch, J.; Brisson, N. Crystal structures of DNA-Whirly complexes and their role in Arabidopsis organelle genome repair. Plant Cell 2010, 22, 1849–1867. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Chen, D.; Teng, L.; Guan, P.; Yu, G.; Zhang, P.; Song, J.; Zeng, Q.; Zhu, L. OsWHY1 interacts with OsTRX z and is essential for early chloroplast development in rice. Rice 2022, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Schwacke, R.; Fischer, K.; Ketelsen, B.; Krupinska, K.; Krause, K. Comparative survey of plastid and mitochondrial targeting properties of transcription factors in Arabidopsis and rice. Mol. Genet. Genom. 2007, 277, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Desveaux, D.; Després, C.; Joyeux, A.; Subramaniam, R.; Brisson, N. PBF-2 is a novel single-stranded DNA binding factor implicated in PR-10a gene activation in potato. Plant Cell 2000, 12, 1477–1489. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Wang, Y.; Xu, H.; Wang, B.; Zhu, X.; Wei, B.; Wei, X. Genome-wide identification, phylogenetic, and expression analysis under abiotic stress conditions of Whirly (WHY) gene family in Medicago sativa L. Sci. Rep. 2022, 12, 18676. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, D.; Shi, X.; Deng, B.; Ren, Y.; Lin, W.; Miao, Y. H2O2 as a feedback signal on dual-located WHIRLY1 associates with leaf senescence in Arabidopsis. Cells 2019, 8, 1585. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, K.; Yang, Z.; Hou, Q.; Zhao, W.W.; Sun, Q. RNase H1C collaborates with ssDNA binding proteins WHY1/3 and recombinase RecA1 to fulfill the DNA damage repair in Arabidopsis chloroplasts. Nucleic Acids Res. 2021, 49, 6771–6787. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhang, H.; Huang, D.; Schenke, D.; Cai, D.; Wu, B.; Miao, Y. Dual-localized WHIRLY1 affects salicylic acid biosynthesis via coordination of ISOCHORISMATE SYNTHASE1, PHENYLALANINE AMMONIA LYASE1, and S-ADENOSYL-L-METHIONINE-DEPENDENT METHYLTRANSFERASE1. Plant Physiol. 2020, 184, 1884–1899. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Wang, J.; Jiao, B.; Chen, C.; Zhang, J.; Ma, N.; Meng, Q. WHIRLY1 maintains leaf photosynthetic capacity in tomato by regulating the expression of RbcS1 under chilling stress. J. Exp. Bot. 2020, 71, 3653–3663. [Google Scholar] [CrossRef]
- Zhuang, K.; Gao, Y.; Liu, Z.; Diao, P.; Sui, N.; Meng, Q.; Meng, C.; Kong, F. WHIRLY1 regulates HSP21.5A expression to promote thermotolerance in tomato. Plant Cell Physiol. 2020, 61, 169–177. [Google Scholar] [CrossRef]
- Guan, Z.; Wang, W.; Yu, X.; Lin, W.; Miao, Y. Comparative proteomic analysis of coregulation of CIPK14 and WHIRLY1/3 mediated pale yellowing of leaves in Arabidopsis. Int. J. Mol. Sci. 2018, 19, 2231. [Google Scholar] [CrossRef]
- Huang, C.; Yu, J.; Cai, Q.; Chen, Y.; Li, Y.; Ren, Y.; Miao, Y. Triple-localized WHIRLY2 influences leaf senescence and silique development via carbon allocation. Plant Physiol. 2020, 184, 1348–1362. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011, 11, 136. [Google Scholar] [CrossRef]
- Reiser, L.; Subramaniam, S.; Li, D.; Huala, E. Using the Arabidopsis information resource (TAIR) to find information about Arabidopsis genes. Curr. Protoc. Bioinform. 2017, 60, e574. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Liu, D.; Yu, L.; Wei, L.; Yu, P.; Wang, J.; Zhao, H.; Zhang, Y.; Zhang, S.; Yang, Z.; Chen, G.; et al. BnTIR: An online transcriptome platform for exploring RNA-seq libraries for oil crop Brassica napus. Plant Biotechnol. J. 2021, 19, 1895–1897. [Google Scholar] [CrossRef]
- Desveaux, D.; Allard, J.; Brisson, N.; Sygusch, J. A new family of plant transcription factors displays a novel ssDNA-binding surface. Nat. Struct. Biol. 2002, 9, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome size diversity and its impact on the evolution of land plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Han, Y.; Meng, D.; Li, D.; Jin, Q.; Lin, Y.; Cai, Y. Structural, evolutionary, and functional analysis of the class III peroxidase gene family in Chinese pear (Pyrus bretschneideri). Front. Plant Sci. 2016, 7, 1874. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Evolution: Polyploid gains. Nat. Rev. Genet. 2015, 16, 196. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Inatsugi, R.; Terada, A.; Hirose, K.; Kudoh, H.; Sese, J.; Shimizu, K.K. Plant adaptive radiation mediated by polyploid plasticity in transcriptomes. Mol. Ecol. 2017, 26, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef]
- Nuruzzaman, M.; Sharoni, A.M.; Satoh, K.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative transcriptome profiles of the WRKY gene family under control, hormone-treated, and drought conditions in near-isogenic rice lines reveal differential, tissue specific gene activation. J. Plant Physiol. 2014, 171, 2–13. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Chromosome | Number of Amino Acid (aa) | Molecular Weight (Da) | Isoelectric Point (pI) | Instability Index | Orthologous Gene |
|---|---|---|---|---|---|---|---|
| BrWHY1a | BraA06g039670.3C | A06 | 261 | 28,804.77 | 9.56 | 39.52 | AT2G02740 |
| BrWHY2a | BraA07g035880.3C | A07 | 240 | 26,097.76 | 9.46 | 47.03 | AT1G71260 |
| BrWHY3a | BraA08g030230.3C | A08 | 269 | 29,404.12 | 9.30 | 47.37 | AT1G14410 |
| BoWHY1a | BolC06g042680.2J | C06 | 238 | 26,120.87 | 9.48 | 43.55 | AT1G71260 |
| BoWHY2a | BolC07g032390.2J | C07 | 261 | 28,763.65 | 9.58 | 44.89 | AT2G02740 |
| BoWHY3a | BolC08g025140.2J | C08 | 267 | 29,185.91 | 9.30 | 45.73 | AT1G14410 |
| BoWHY4 | BolC07g016070.2J | C07 | 155 | 17,246.77 | 10.22 | 39.28 | AT1G14410 |
| BnAWHY1b | BnaA06G0401800ZS | chrA06 | 129 | 14,290.28 | 6.10 | 27.58 | AT2G02740 |
| BnAWHY2b | BnaA07G0324700ZS | chrA07 | 239 | 26,033.72 | 9.46 | 48.24 | AT1G71260 |
| BnAWHY3b | BnaA08G0272100ZS | chrA08 | 95 | 10,643.99 | 4.91 | 40.17 | AT1G14410 |
| BnCWHY1b | BnaC06G0379900ZS | chrC06 | 250 | 27,481.34 | 9.18 | 41.95 | AT1G71260 |
| BnCWHY2b | BnaC07G0279600ZS | chrC07 | 260 | 28,635.52 | 9.58 | 45.02 | AT2G02740 |
| BnCWHY3b | BnaC08G0228600ZS | chrC08 | 267 | 29,225.93 | 9.30 | 44.37 | AT1G14410 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhao, Z.; Li, H.; Pei, D.; Ma, Q.; Huang, Z.; Wang, H.; Xiao, L. Genome-Wide Identification and Molecular Evolutionary History of the Whirly Family Genes in Brassica napus. Plants 2024, 13, 2243. https://doi.org/10.3390/plants13162243
Wang L, Zhao Z, Li H, Pei D, Ma Q, Huang Z, Wang H, Xiao L. Genome-Wide Identification and Molecular Evolutionary History of the Whirly Family Genes in Brassica napus. Plants. 2024; 13(16):2243. https://doi.org/10.3390/plants13162243
Chicago/Turabian StyleWang, Long, Zhi Zhao, Huaxin Li, Damei Pei, Qianru Ma, Zhen Huang, Hongyan Wang, and Lu Xiao. 2024. "Genome-Wide Identification and Molecular Evolutionary History of the Whirly Family Genes in Brassica napus" Plants 13, no. 16: 2243. https://doi.org/10.3390/plants13162243