




Pharmaceutics 2024, 16(12), 1617; https://doi.org/10.3390/pharmaceutics16121617 - 20 Dec 2024
Viewed by 1056
Abstract
►
Show Figures
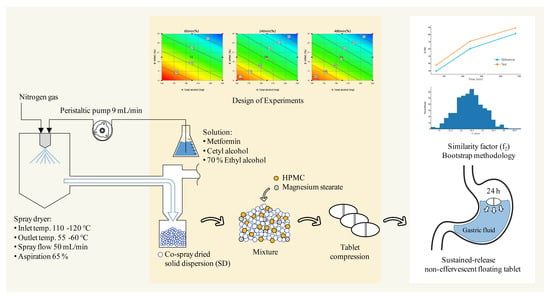
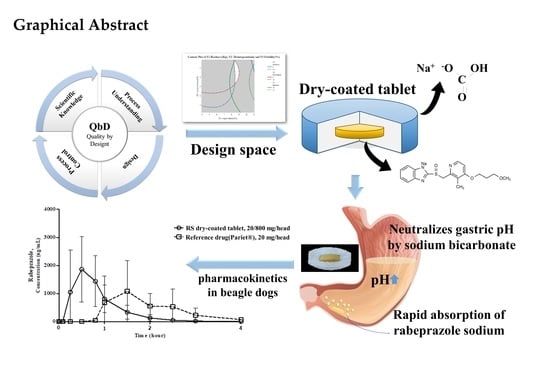
Background/Objectives: A sustained-release formulation of fenofibrate while enhancing drug dissolution with minimal food effect is critical for maximizing the therapeutic benefits of fenofibrate. Therefore, this study aimed to develop an effective solid dispersion formulation of fenofibrate for simultaneous enhancement in the extent and
[...] Read more.
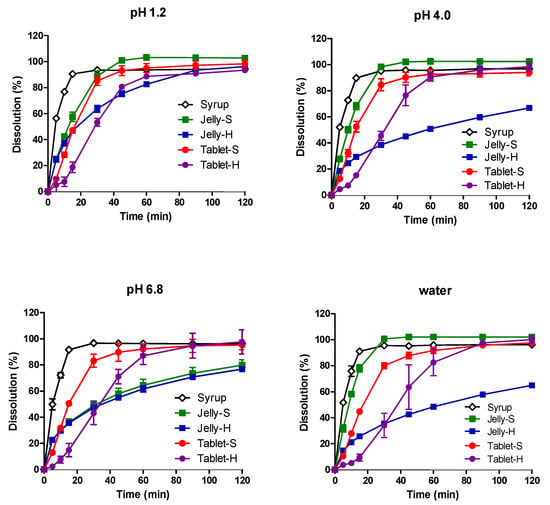
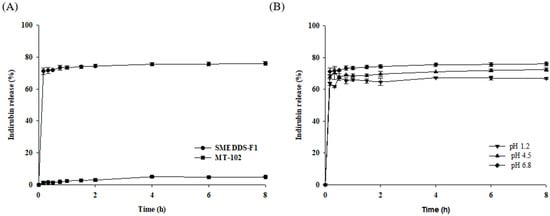
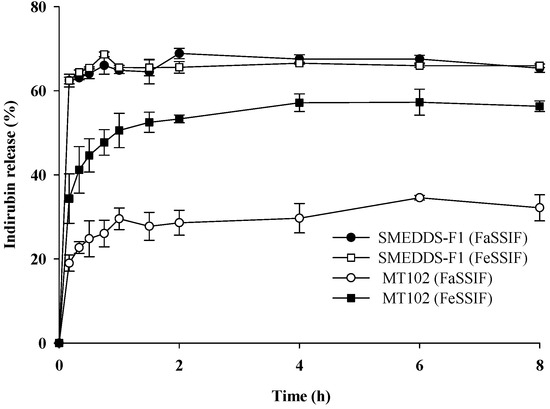
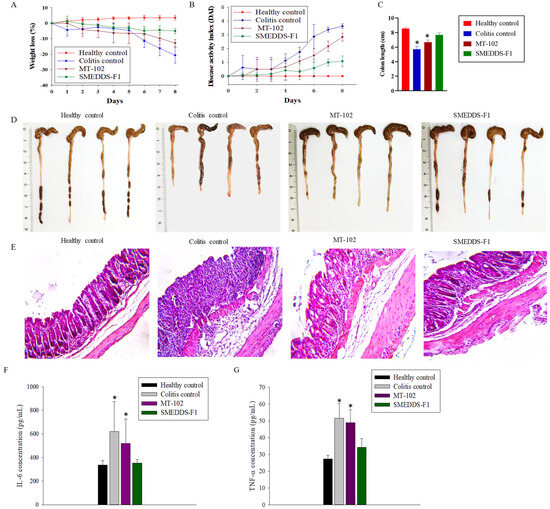
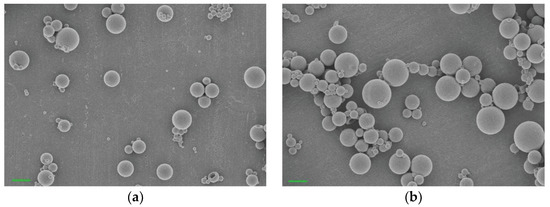
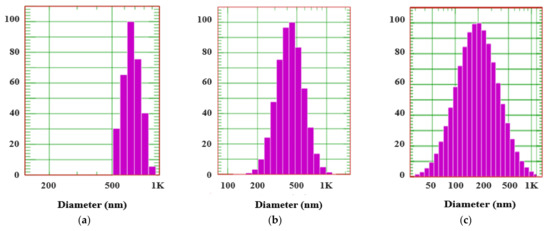
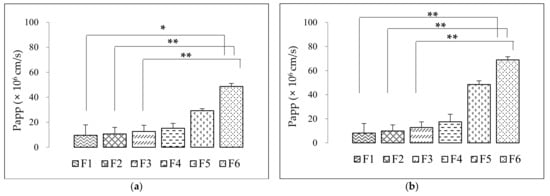
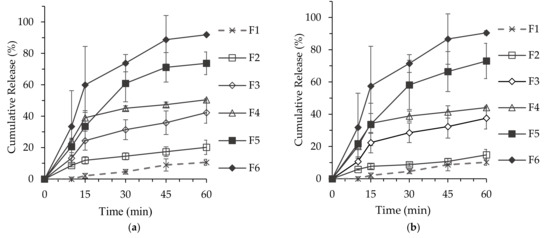
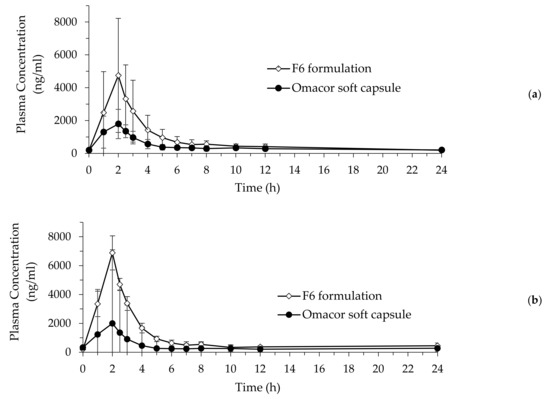
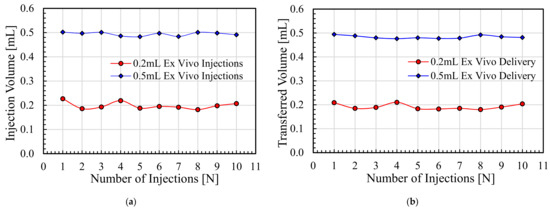
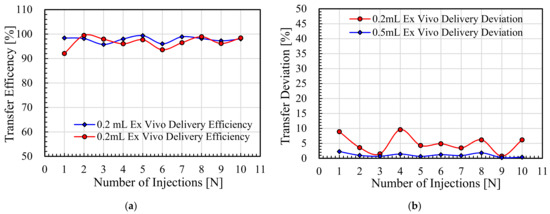
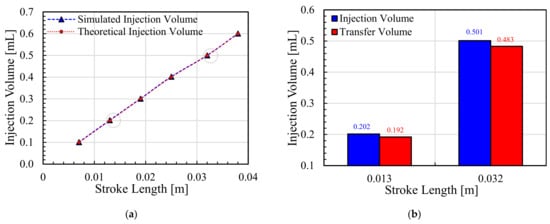
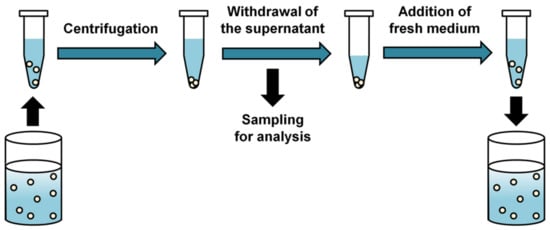
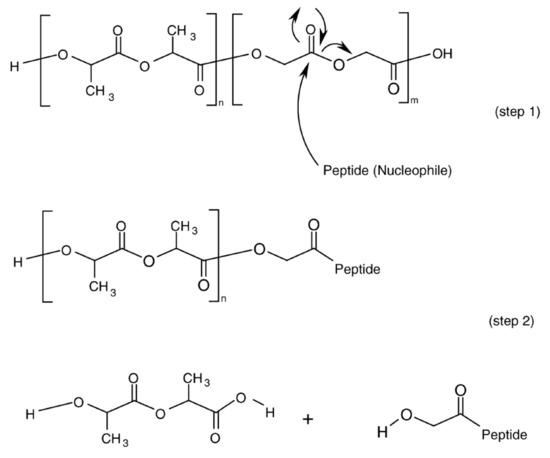
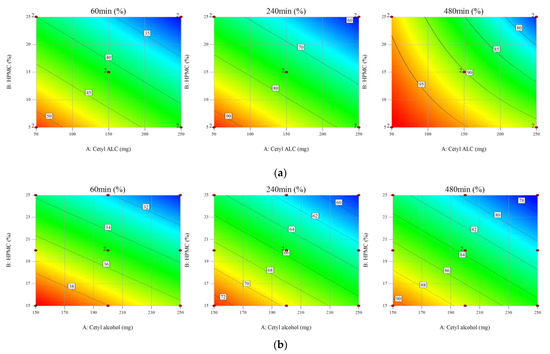
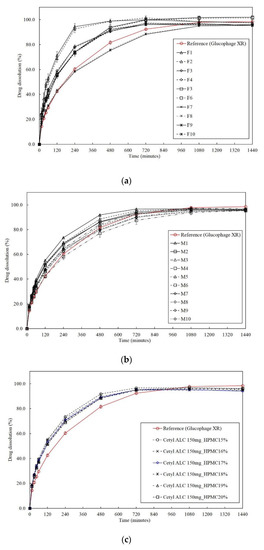
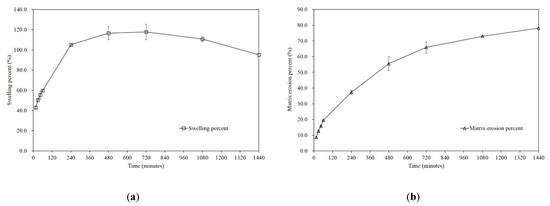
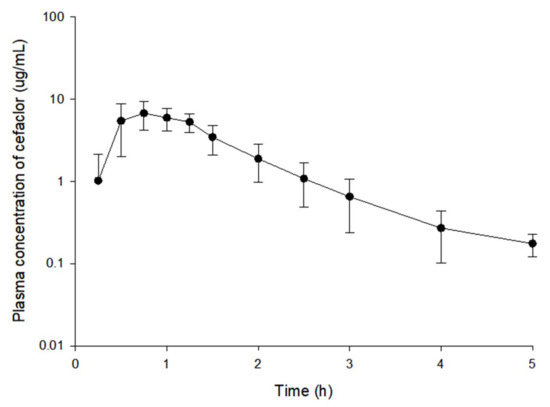
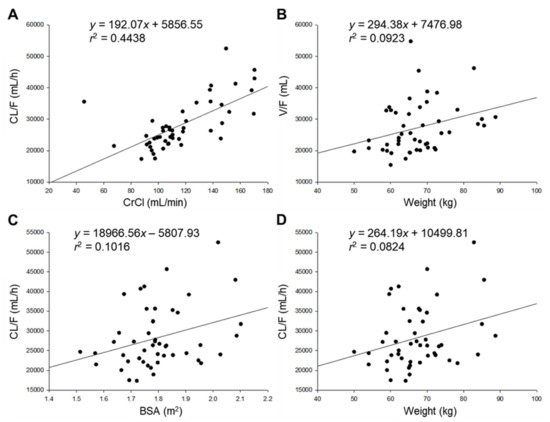
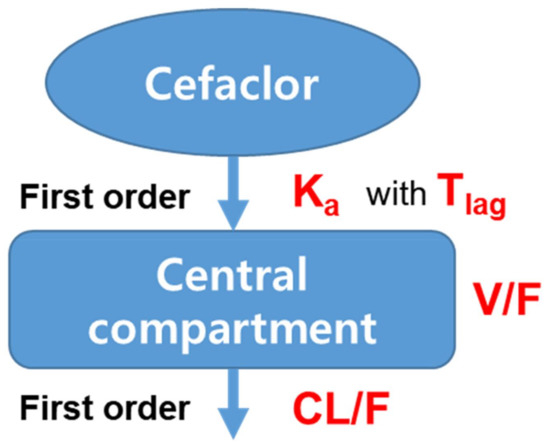
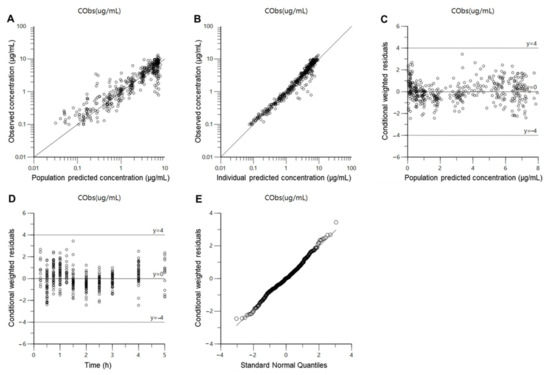
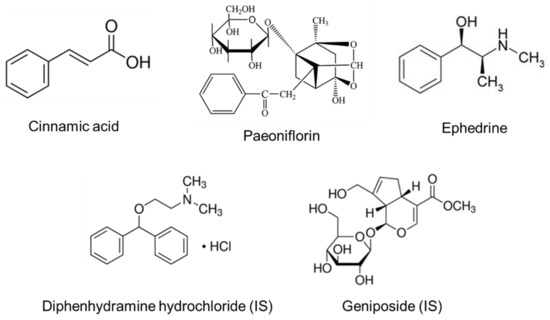
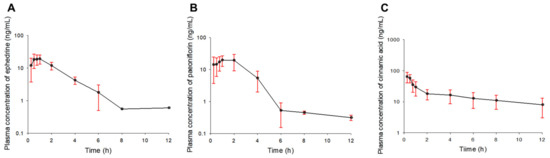
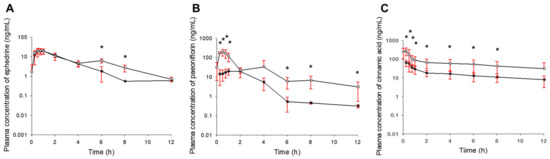
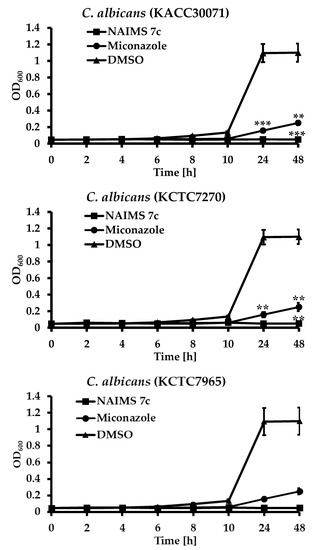
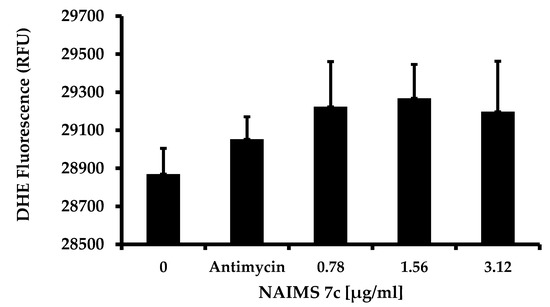
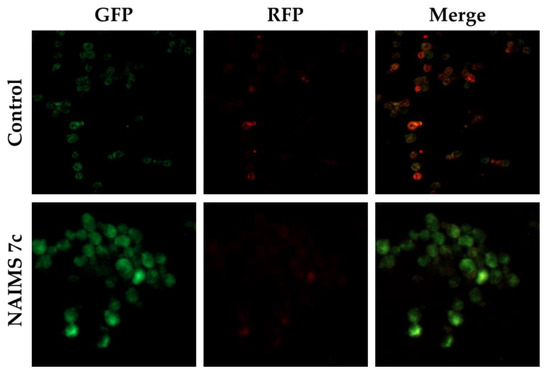
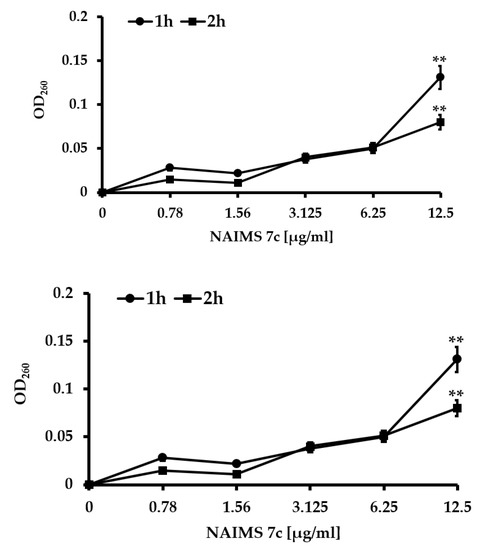
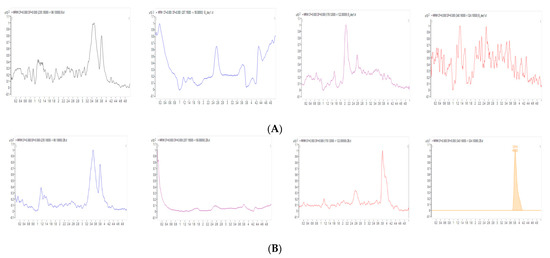
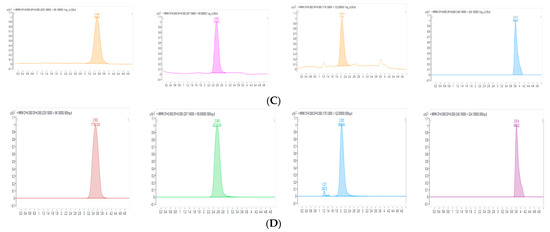
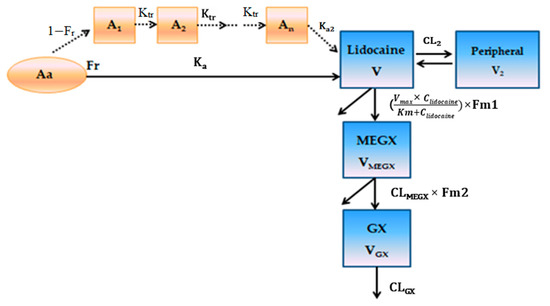
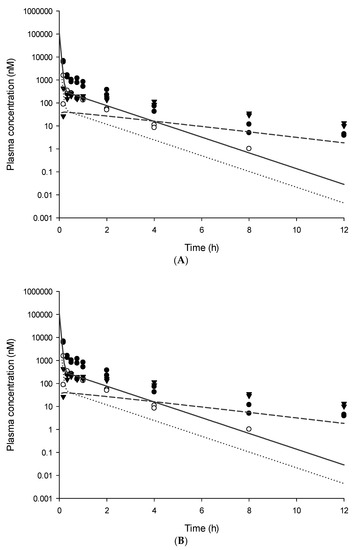
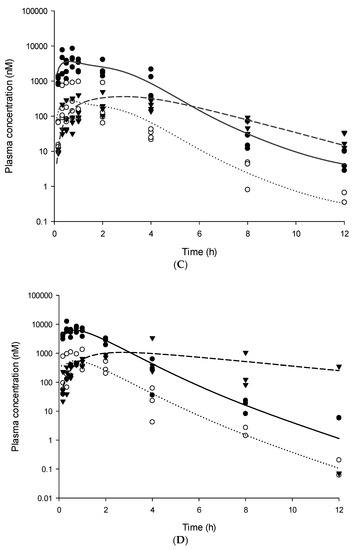
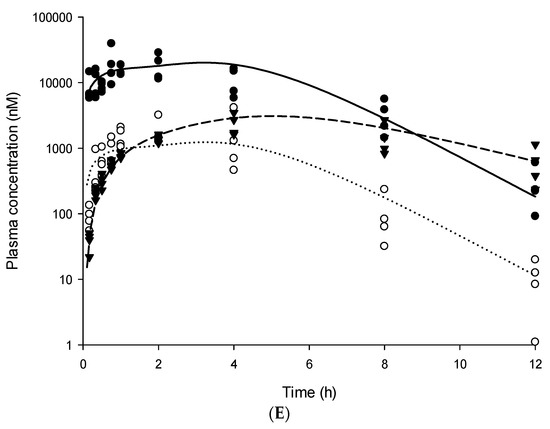
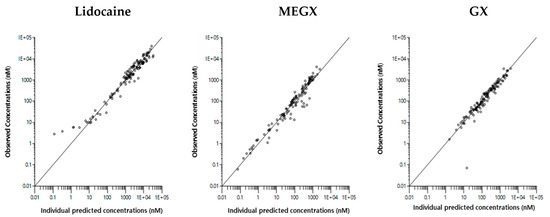
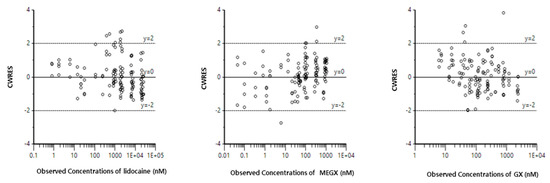
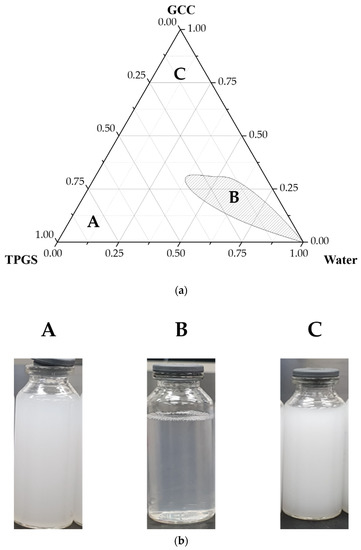
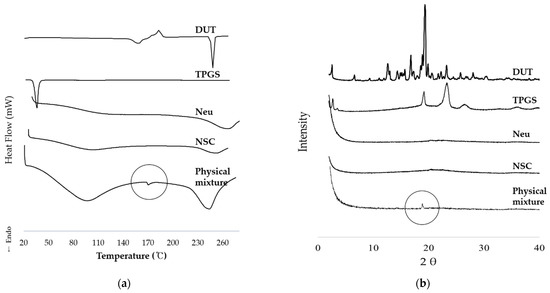
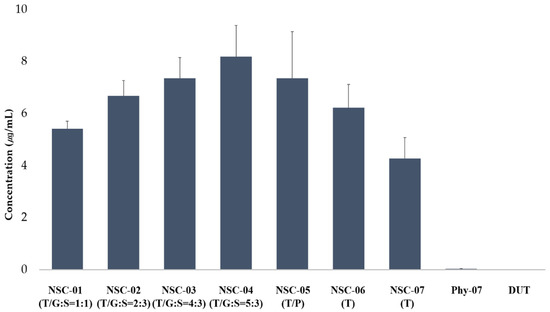
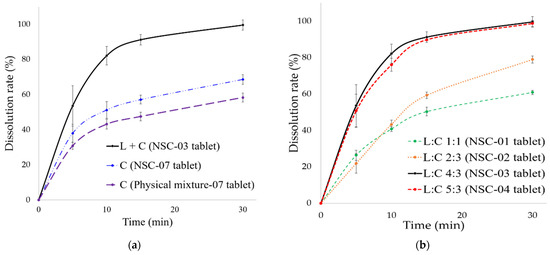
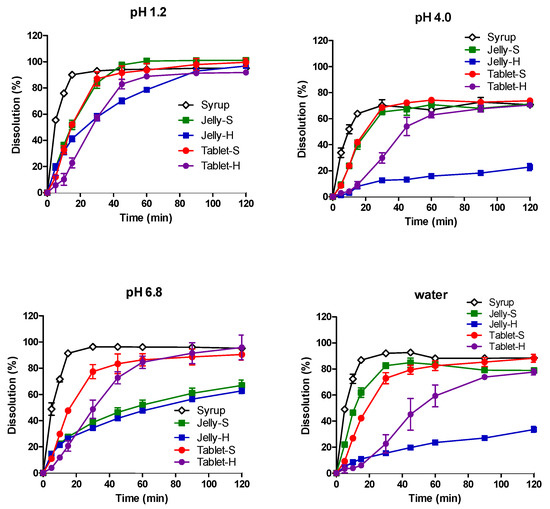
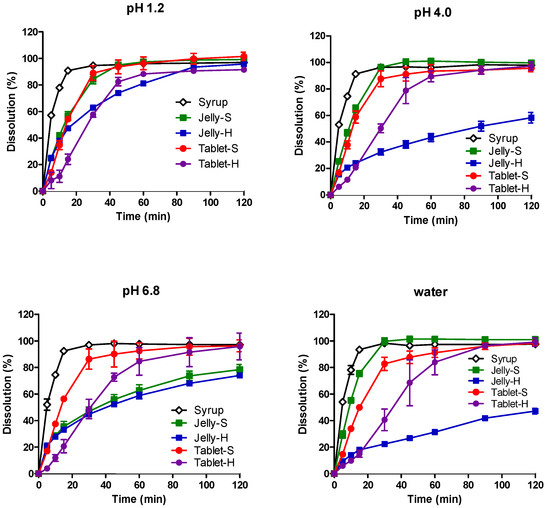
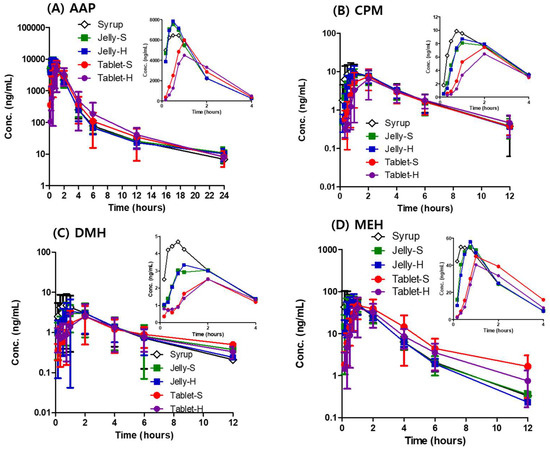
Background/Objectives: A sustained-release formulation of fenofibrate while enhancing drug dissolution with minimal food effect is critical for maximizing the therapeutic benefits of fenofibrate. Therefore, this study aimed to develop an effective solid dispersion formulation of fenofibrate for simultaneous enhancement in the extent and duration of drug exposure. Methods: Fenofibrate-loaded solid dispersions (FNSDs) were prepared using poloxamer 407 and Eudragit® RSPO at varied ratios via solvent evaporation. In vitro/in vivo characteristics of FNSDs were examined in comparison with untreated drugs. Results: Based on dissolution profiles of FNSDs in aqueous media, the weight ratio of fenofibrate: poloxamer 407: Eudragit® RSPO at 1:1:4 (FNSD2) was selected as the optimal composition for achieving sustained drug release while maximizing the drug dissolution. The enhanced and sustained drug release of FNSD2 was also confirmed in a buffer transition system mimicking the pH change in the gastrointestinal tract. FNSD2 achieved approximately 66% drug release over 12 h, while pure drug exhibited only 12%. Furthermore, FNSD2 maintained similar release rates under fed and fasted conditions, while the entire drug dissolution slightly increased in the fed state. Structural analysis by x-ray diffraction showed that fenofibrate remained crystalline in FNSD2. Pharmacokinetic studies in rats revealed that orally administered FNSD2 significantly improved the extent and duration of systemic drug exposure. Compared to pure drugs, the FNSD2 formulation increased the oral bioavailability of fenofibrate by 22 folds with the delayed Tmax of 4 h in rats. Conclusion: FNSD2 formulation is effective in improving the extent and duration of drug exposure simultaneously.
Full article

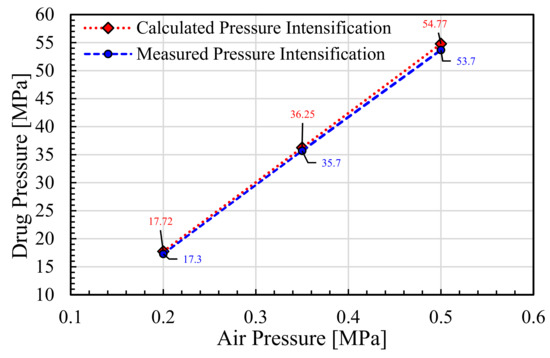
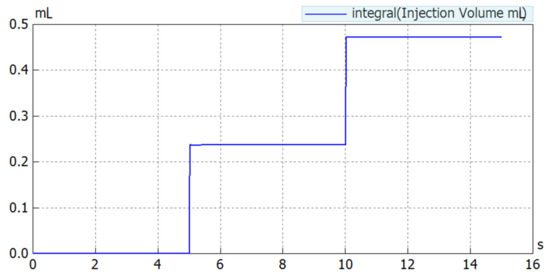
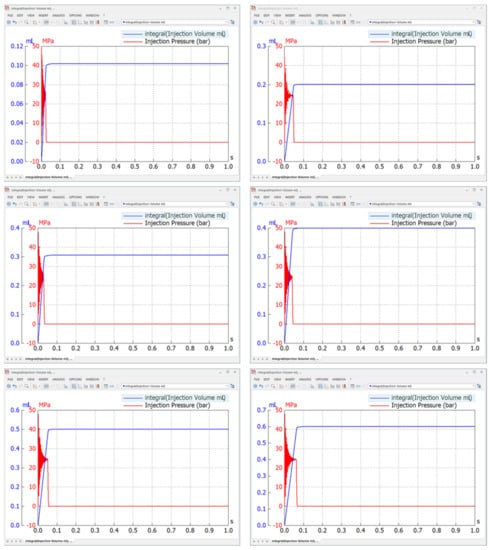
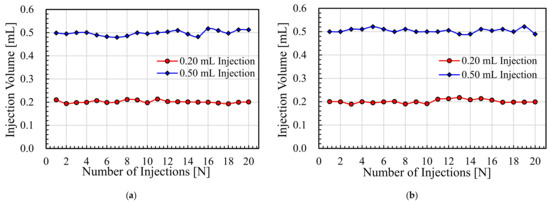
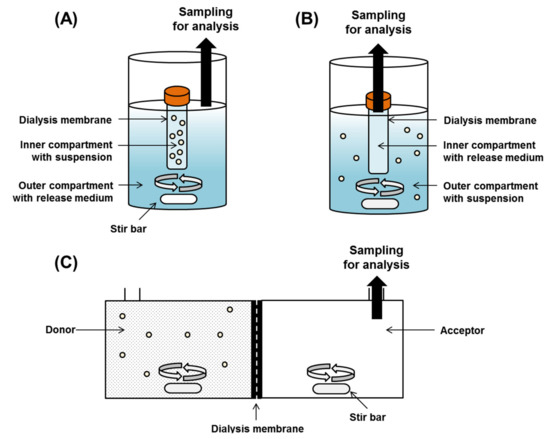
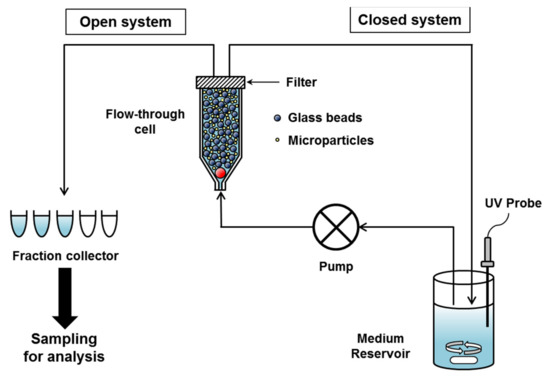
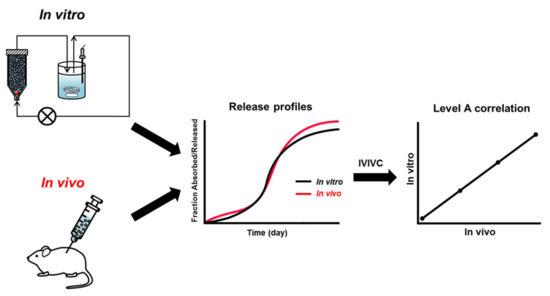
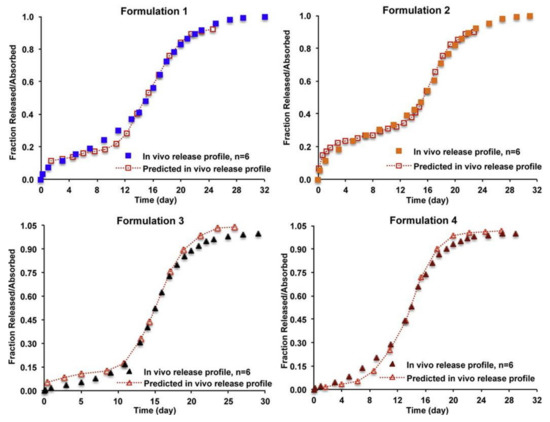
Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}