
The Transcriptional Landscape of Berry Skin in Red and White PIWI (“Pilzwiderstandsfähig”) Grapevines Possessing QTLs for Partial Resistance to Downy and Powdery Mildews
 , , ,
, , ,  , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
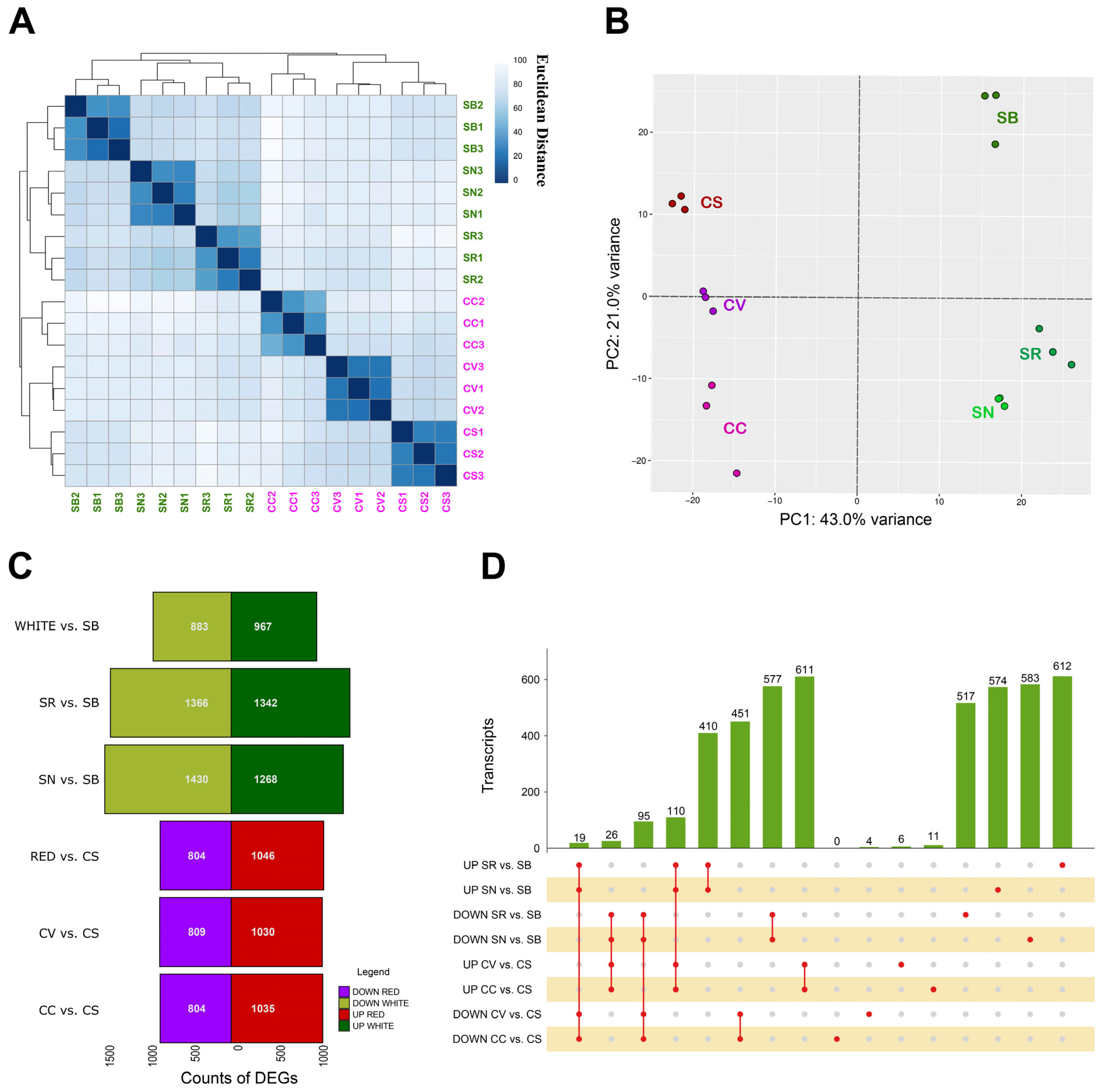
2.1. Global Transcriptome Analysis of PIWI and Elite Varieties and Identification of Differentially Expressed Genes in Tolerant vs. Susceptible Red and White Varieties
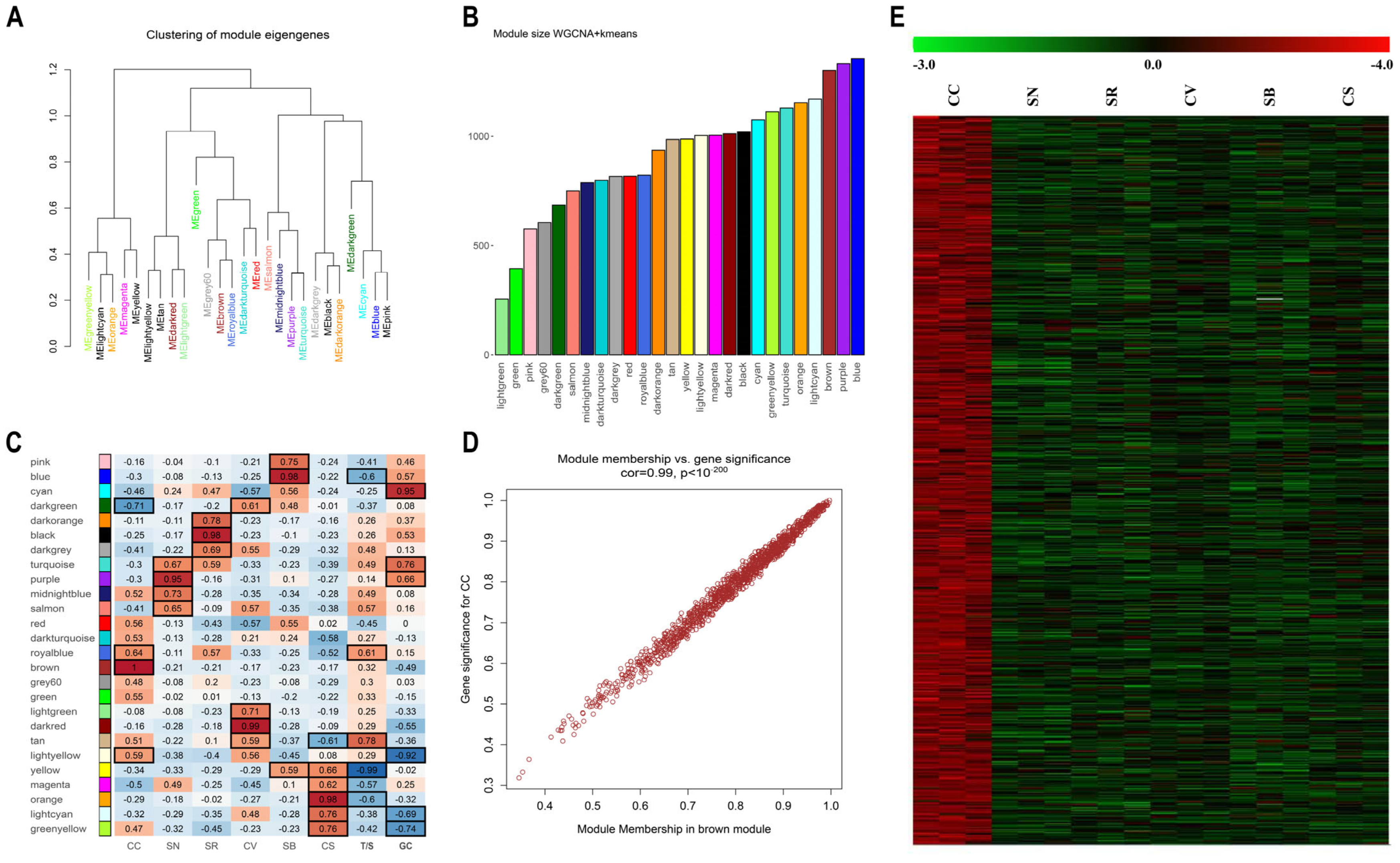
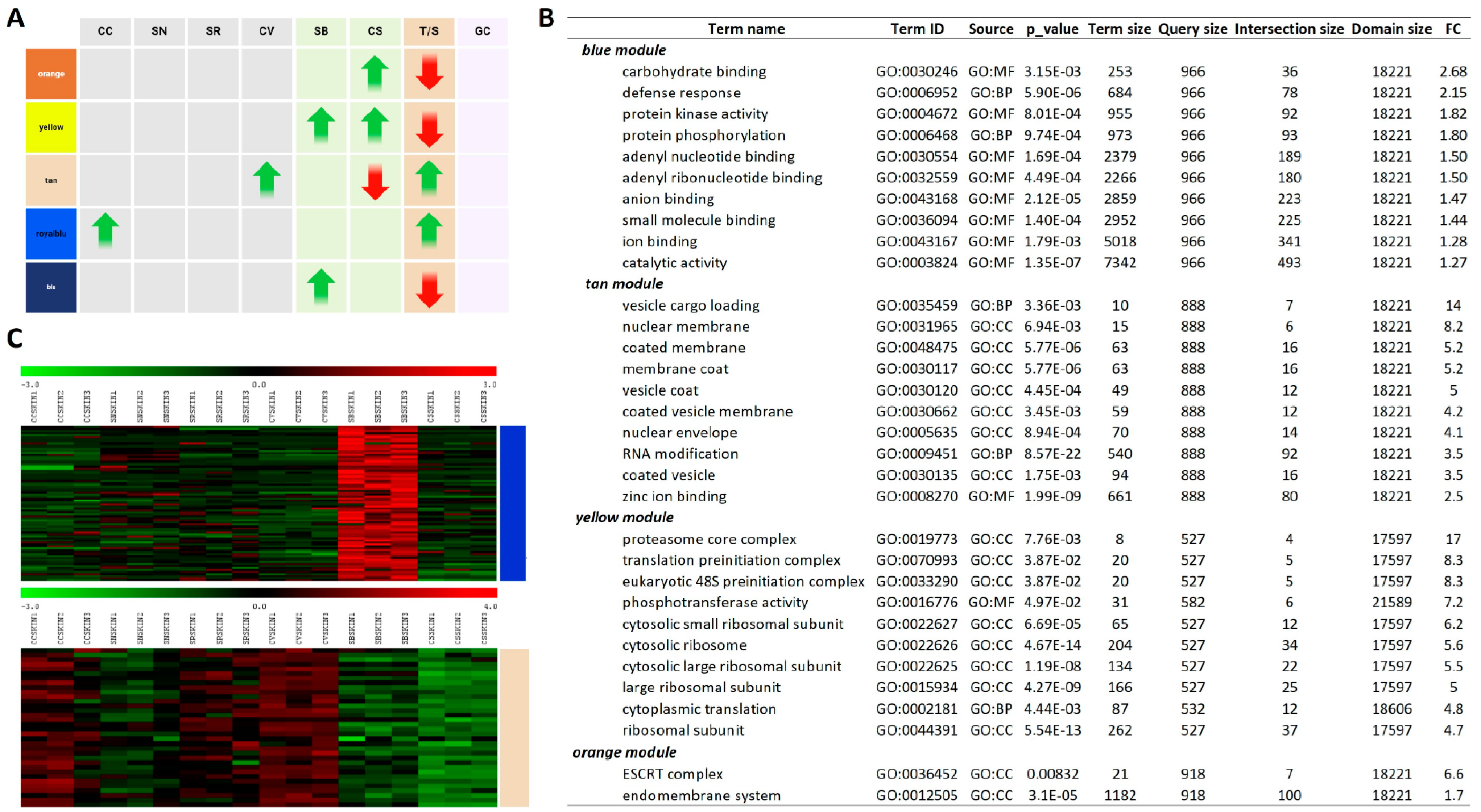
2.2. K-Means Associated Weighted Gene Co-Expression Network Analysis Indicates Gene Clusters Associated with PIWI Genotypes and Tolerance
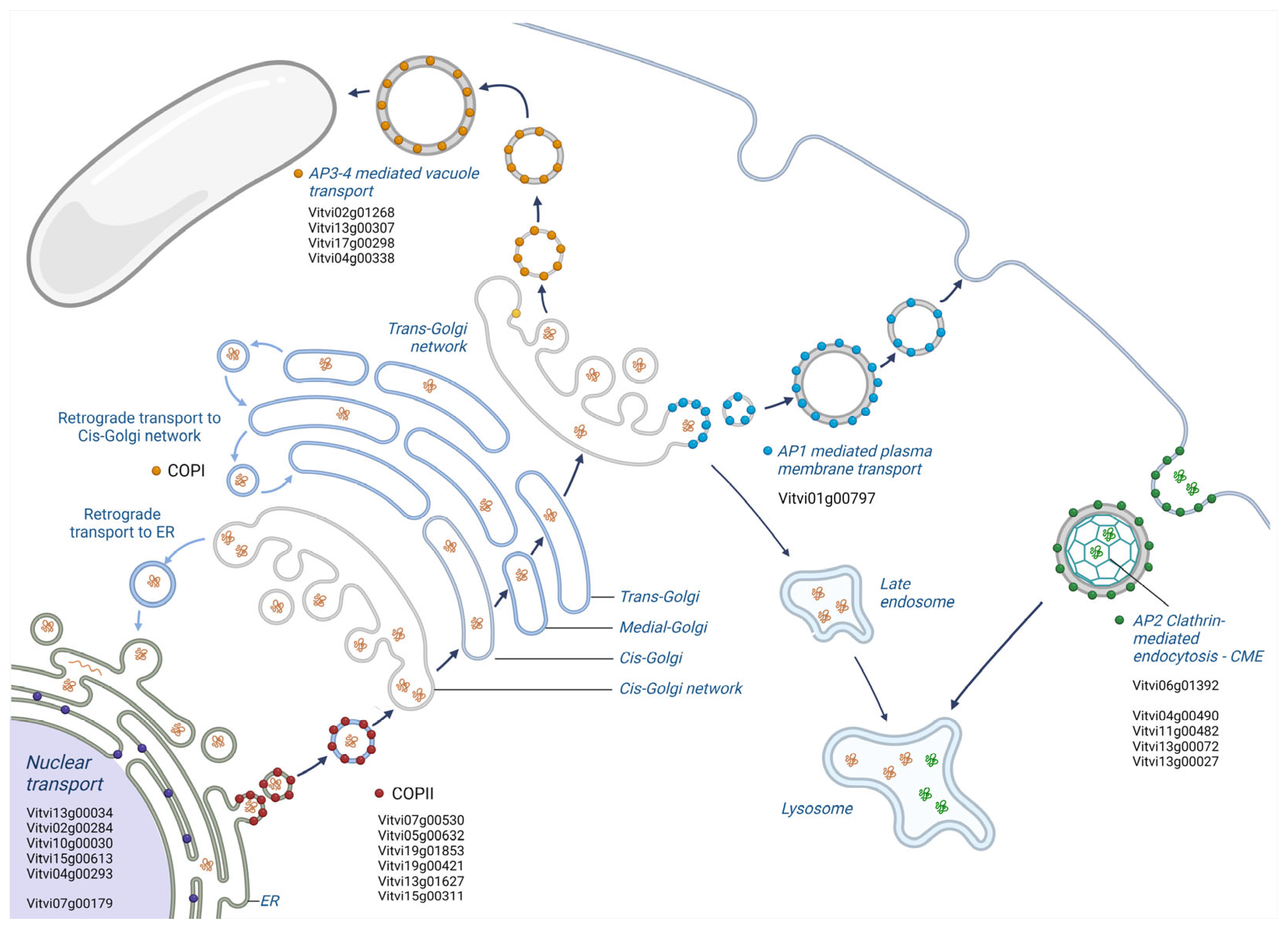
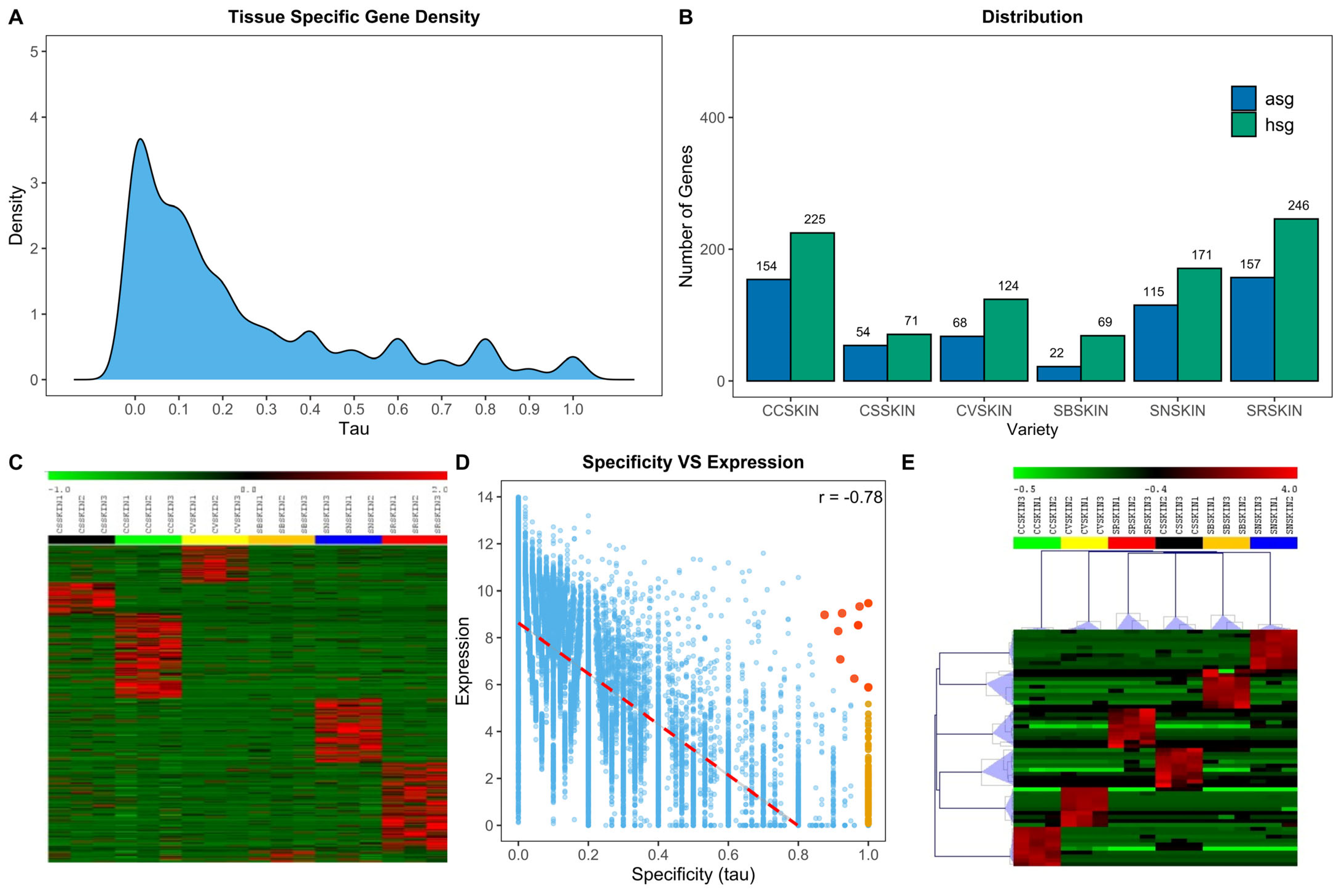
2.3. PIWI Varieties Show Absolutely and Highly Specific Genes Which Are Involved in Disease Resistance
3. Materials and Methods
3.1. Plant Material and Sample Collection
3.2. RNA Purification, Library Preparation, and Sequencing
3.3. RNA-seq Analysis
3.4. Differential Gene Expression Analysis
3.5. K-Means Corrected Weighted Gene Co-Expression Network Analysis
3.6. Identification of Highly Specific Genes (HSG) and Absolutely Specific Genes (ASG) by Means of Tau Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guzmán-Ardiles, R.E.; Pegoraro, C.; da Maia, L.C.; Costa de Oliveira, A. Genetic Changes in the Genus Vitis and the Domestication of Vine. Front. Plant Sci. 2022, 13, 1019311. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; Arroyo-Garcia, R. Editorial: Origins and Domestication of the Grape. Front. Plant Sci. 2020, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Forneck, A.; Walker, M.A.; Blaich, R.; Yvon, M.; Leclant, F. Interaction of Phylloxera (Daktulosphaira Vitifoliae Fitch) with Grape (Vitis Spp.) in Simple Isolation Chambers. Am. J. Enol. Vitic. 2001, 52, 28–34. [Google Scholar] [CrossRef]
- Gessler, C.; Pertot, I.; Perazzolli, M. Plasmopara viticola: A Review of Knowledge on Downy Mildew of Grapevine and Effective Disease Management. Phytopathol. Mediterr. 2011, 50, 3–44. [Google Scholar]
- Gadoury, D.M.; Cadle-Davidson, L.; Wilcox, W.F.; Dry, I.B.; Seem, R.C.; Milgroom, M.G. Grapevine Powdery Mildew (Erysiphe Necator): A Fascinating System for the Study of the Biology, Ecology and Epidemiology of an Obligate Biotroph. Mol. Plant Pathol. 2012, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Caffi, T.; Gilardi, G.; Monchiero, M.; Rossi, V. Production and Release of Asexual Sporangia in Plasmopara viticola. Phytopathology 2013, 103, 64–73. [Google Scholar] [CrossRef]
- Mouafo-Tchinda, R.A.; Beaulieu, C.; Fall, M.L.; Carisse, O. Effect of Temperature on Aggressiveness of Plasmopara Viticola f. Sp. Aestivalis and P. Viticola f. Sp. Riparia from Eastern Canada. Can. J. Plant Pathol. 2021, 43, 73–87. [Google Scholar] [CrossRef]
- Carroll, J.E.; Wilcox, W.F. Effects of Humidity on the Development of Grapevine Powdery Mildew. Phytopathology 2003, 93, 1137–1144. [Google Scholar] [CrossRef]
- Nicolopoulou-Stamati, P.; Maipas, S.; Kotampasi, C.; Stamatis, P.; Hens, L. Chemical Pesticides and Human Health: The Urgent Need for a New Concept in Agriculture. Front. Public Health 2016, 4, 148. [Google Scholar] [CrossRef]
- Possamai, T.; Wiedemann-Merdinoglu, S. Phenotyping for QTL Identification: A Case Study of Resistance to Plasmopara Viticola and Erysiphe Necator in Grapevine. Front. Plant Sci. 2022, 13, 930954. [Google Scholar] [CrossRef]
- Marguerit, E.; Boury, C.; Manicki, A.; Donnart, M.; Butterlin, G.; Némorin, A.; Wiedemann-Merdinoglu, S.; Merdinoglu, D.; Ollat, N.; Decroocq, S. Genetic Dissection of Sex Determinism, Inflorescence Morphology and Downy Mildew Resistance in Grapevine. Theor. Appl. Genet. 2009, 118, 1261–1278. [Google Scholar] [CrossRef] [PubMed]
- Pap, D.; Riaz, S.; Dry, I.B.; Jermakow, A.; Tenscher, A.C.; Cantu, D.; Oláh, R.; Walker, M.A. Identification of Two Novel Powdery Mildew Resistance Loci, Ren6 and Ren7, from the Wild Chinese Grape Species Vitis Piasezkii. BMC Plant Biol. 2016, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, S.; Chen, L.-L.; Yang, S.; Hyma, K.E.; Cadle-Davidson, L.; Hwang, C.-F. Construction of a High-Density Linkage Map and QTL Detection of Downy Mildew Resistance in Vitis Aestivalis-Derived ‘Norton’. Theor. Appl. Genet. 2019, 132, 137–147. [Google Scholar] [CrossRef]
- Lin, H.; Leng, H.; Guo, Y.; Kondo, S.; Zhao, Y.; Shi, G.; Guo, X. QTLs and Candidate Genes for Downy Mildew Resistance Conferred by Interspecific Grape (V. Vinifera L. × V. Amurensis Rupr.) Crossing. Sci. Hortic. 2019, 244, 200–207. [Google Scholar] [CrossRef]
- Pauquet, J.; Bouquet, A.; This, P.; Adam-Blondon, A.-F. Establishment of a Local Map of AFLP Markers around the Powdery Mildew Resistance Gene Run1 in Grapevine and Assessment of Their Usefulness for Marker Assisted Selection. Theor. Appl. Genet. 2001, 103, 1201–1210. [Google Scholar] [CrossRef]
- Riaz, S.; Tenscher, A.C.; Ramming, D.W.; Walker, M.A. Using a Limited Mapping Strategy to Identify Major QTLs for Resistance to Grapevine Powdery Mildew (Erysiphe Necator) and Their Use in Marker-Assisted Breeding. Theor. Appl. Genet. 2011, 122, 1059–1073. [Google Scholar] [CrossRef]
- Mahanil, S.; Ramming, D.; Cadle-Davidson, M.; Owens, C.; Garris, A.; Myles, S.; Cadle-Davidson, L. Development of Marker Sets Useful in the Early Selection of Ren4 Powdery Mildew Resistance and Seedlessness for Table and Raisin Grape Breeding. Theor. Appl. Genet. 2012, 124, 23–33. [Google Scholar] [CrossRef]
- Karn, A.; Zou, C.; Brooks, S.; Fresnedo-Ramírez, J.; Gabler, F.; Sun, Q.; Ramming, D.; Naegele, R.; Ledbetter, C.; Cadle-Davidson, L. Discovery of the REN11 Locus From Vitis Aestivalis for Stable Resistance to Grapevine Powdery Mildew in a Family Segregating for Several Unstable and Tissue-Specific Quantitative Resistance Loci. Front. Plant Sci. 2021, 12, 733899. [Google Scholar] [CrossRef]
- Welter, L.J.; Göktürk-Baydar, N.; Akkurt, M.; Maul, E.; Eibach, R.; Töpfer, R.; Zyprian, E.M. Genetic Mapping and Localization of Quantitative Trait Loci Affecting Fungal Disease Resistance and Leaf Morphology in Grapevine (Vitis vinifera L.). Mol. Breed. 2007, 20, 359–374. [Google Scholar] [CrossRef]
- Bellin, D.; Peressotti, E.; Merdinoglu, D.; Wiedemann-Merdinoglu, S.; Adam-Blondon, A.-F.; Cipriani, G.; Morgante, M.; Testolin, R.; Di Gaspero, G. Resistance to Plasmopara Viticola in Grapevine ‘Bianca’ Is Controlled by a Major Dominant Gene Causing Localised Necrosis at the Infection Site. Theor. Appl. Genet. 2009, 120, 163–176. [Google Scholar] [CrossRef]
- Venuti, S.; Copetti, D.; Foria, S.; Falginella, L.; Hoffmann, S.; Bellin, D.; Cindrić, P.; Kozma, P.; Scalabrin, S.; Morgante, M.; et al. Historical Introgression of the Downy Mildew Resistance Gene Rpv12 from the Asian Species Vitis Amurensis into Grapevine Varieties. PLoS ONE 2013, 8, e61228. [Google Scholar] [CrossRef] [PubMed]
- Vezzulli, S.; Malacarne, G.; Masuero, D.; Vecchione, A.; Dolzani, C.; Goremykin, V.; Mehari, Z.H.; Banchi, E.; Velasco, R.; Stefanini, M.; et al. The Rpv3-3 Haplotype and Stilbenoid Induction Mediate Downy Mildew Resistance in a Grapevine Interspecific Population. Front. Plant Sci. 2019, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, V.; Crespan, M.; Maddalena, G.; Migliaro, D.; Brancadoro, L.; Maghradze, D.; Failla, O.; Toffolatti, S.L.; De Lorenzis, G. Novel Loci Associated with Resistance to Downy and Powdery Mildew in Grapevine. Front. Plant Sci. 2024, 15, 1386225. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei, M.; Maddalena, G.; Bitsadze, N.; Maghradze, D.; Bianco, P.A.; Failla, O.; Toffolatti, S.L.; De Lorenzis, G. Rpv29, Rpv30 and Rpv31: Three Novel Genomic Loci Associated With Resistance to Plasmopara Viticola in Vitis Vinifera. Front. Plant Sci. 2020, 11, 562432. [Google Scholar] [CrossRef] [PubMed]
- Di Gaspero, G.; Copetti, D.; Coleman, C.; Castellarin, S.D.; Eibach, R.; Kozma, P.; Lacombe, T.; Gambetta, G.; Zvyagin, A.; Cindrić, P.; et al. Selective Sweep at the Rpv3 Locus during Grapevine Breeding for Downy Mildew Resistance. Theor. Appl. Genet. 2012, 124, 277–286. [Google Scholar] [CrossRef]
- Peressotti, E.; Dolzani, C.; Poles, L.; Banchi, E.; Stefanini, M.; Salamini, F.; Velasco, R.; Vezzulli, S.; Riaz, S.; Walker, M.A.; et al. A first pedigree-based analysis (PBA) approach for the dissection of disease resistance traits in grapevine hybrids. Acta Hortic. 2015, 113–121. [Google Scholar] [CrossRef]
- Vezzulli, S.; Gramaje, D.; Tello, J.; Gambino, G.; Bettinelli, P.; Pirrello, C.; Schwandner, A.; Barba, P.; Angelini, E.; Anfora, G.; et al. Genomic Designing for Biotic Stress Resistant Grapevine. In Genomic Designing for Biotic Stress Resistant Fruit Crops; Kole, C., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 87–255. ISBN 978-3-030-91801-9. [Google Scholar]
- Tello, J.; Ibáñez, J. Review: Status and Prospects of Association Mapping in Grapevine. Plant Sci. 2023, 327, 111539. [Google Scholar] [CrossRef]
- Pirrello, C.; Magon, G.; Palumbo, F.; Farinati, S.; Lucchin, M.; Barcaccia, G.; Vannozzi, A. Past, Present, and Future of Genetic Strategies to Control Tolerance to the Main Fungal and Oomycete Pathogens of Grapevine. J. Exp. Bot. 2023, 74, 1309–1330. [Google Scholar] [CrossRef]
- Zanghelini, J.A.; Bogo, A.; Dal Vesco, L.L.; Gomes, B.R.; Mecabô, C.V.; Herpich, C.H.; Welter, L.J. Response of PIWI Grapevine Cultivars to Downy Mildew in Highland Region of Southern Brazil. Eur. J. Plant Pathol. 2019, 154, 1051–1058. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Y.; Zhang, H.; Huang, H.; Folta, K.M.; Lu, J. Whole Genome Wide Expression Profiles of Vitis Amurensisgrape Responding to Downy Mildew by Using Solexa Sequencing Technology. BMC Plant Biol. 2010, 10, 234. [Google Scholar] [CrossRef]
- Legay, G.; Marouf, E.; Berger, D.; Neuhaus, J.-M.; Mauch-Mani, B.; Slaughter, A. Identification of Genes Expressed during the Compatible Interaction of Grapevine with Plasmopara Viticola through Suppression Subtractive Hybridization (SSH). Eur. J. Plant Pathol. 2011, 129, 281–301. [Google Scholar] [CrossRef]
- Vannozzi, A.; Dry, I.B.; Fasoli, M.; Zenoni, S.; Lucchin, M. Genome-Wide Analysis of the Grapevine Stilbene Synthase Multigenic Family: Genomic Organization and Expression Profiles upon Biotic and Abiotic Stresses. BMC Plant Biol. 2012, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, D.; Amaro, R.; Erban, A.; Mauri, N.; Soares, F.; Rego, C.; Martínez-Zapater, J.M.; Mithöfer, A.; Kopka, J.; Fortes, A.M. Transcriptional, Hormonal, and Metabolic Changes in Susceptible Grape Berries under Powdery Mildew Infection. J. Exp. Bot. 2021, 72, 6544–6569. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.; Fortes, A.M.; Ferreira, S.; Sebastiana, M.; Choi, Y.H.; Sousa, L.; Acioli-Santos, B.; Pessoa, F.; Verpoorte, R.; Pais, M.S. Transcriptional and Metabolic Profiling of Grape (Vitis vinifera L.) Leaves Unravel Possible Innate Resistance against Pathogenic Fungi. J. Exp. Bot. 2008, 59, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.; Monteiro, F.; Fortes, A.M.; Bonow-Rex, M.; Zyprian, E.; Sousa, L.; Pais, M.S. Cultivar-Specific Kinetics of Gene Induction during Downy Mildew Early Infection in Grapevine. Funct. Integr. Genom. 2012, 12, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Polesani, M.; Bortesi, L.; Ferrarini, A.; Zamboni, A.; Fasoli, M.; Zadra, C.; Lovato, A.; Pezzotti, M.; Delledonne, M.; Polverari, A. General and Species-Specific Transcriptional Responses to Downy Mildew Infection in a Susceptible (Vitis vinifera) and a Resistant (V. riparia) Grapevine Species. BMC Genom. 2010, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Malacarne, G.; Vrhovsek, U.; Zulini, L.; Cestaro, A.; Stefanini, M.; Mattivi, F.; Delledonne, M.; Velasco, R.; Moser, C. Resistance to Plasmopara Viticolain a Grapevine Segregating Population Is Associated with Stilbenoid Accumulation and with Specific Host Transcriptional Responses. BMC Plant Biol. 2011, 11, 114. [Google Scholar] [CrossRef]
- Amrine, K.C.H.; Blanco-Ulate, B.; Riaz, S.; Pap, D.; Jones, L.; Figueroa-Balderas, R.; Walker, M.A.; Cantu, D. Comparative Transcriptomics of Central Asian Vitis Vinifera Accessions Reveals Distinct Defense Strategies against Powdery Mildew. Hortic. Res. 2015, 2, 15037. [Google Scholar] [CrossRef]
- Li, X.; Wu, J.; Yin, L.; Zhang, Y.; Qu, J.; Lu, J. Comparative Transcriptome Analysis Reveals Defense-Related Genes and Pathways against Downy Mildew in Vitis amurensis Grapevine. Plant Physiol. Biochem. 2015, 95, 1–14. [Google Scholar] [CrossRef]
- Eisenmann, B.; Czemmel, S.; Ziegler, T.; Buchholz, G.; Kortekamp, A.; Trapp, O.; Rausch, T.; Dry, I.; Bogs, J. Rpv3–1 Mediated Resistance to Grapevine Downy Mildew Is Associated with Specific Host Transcriptional Responses and the Accumulation of Stilbenes. BMC Plant Biol. 2019, 19, 343. [Google Scholar] [CrossRef]
- Perazzolli, M.; Moretto, M.; Fontana, P.; Ferrarini, A.; Velasco, R.; Moser, C.; Delledonne, M.; Pertot, I. Downy Mildew Resistance Induced by Trichoderma Harzianum T39 in Susceptible Grapevines Partially Mimics Transcriptional Changes of Resistant Genotypes. BMC Genom. 2012, 13, 660. [Google Scholar] [CrossRef] [PubMed]
- Pagliarani, C.; Moine, A.; Chitarra, W.; Meloni, G.R.; Abbà, S.; Nerva, L.; Pugliese, M.; Gullino, M.L.; Gambino, G. The Molecular Priming of Defense Responses Is Differently Regulated in Grapevine Genotypes Following Elicitor Application against Powdery Mildew. Int. J. Mol. Sci. 2020, 21, 6776. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.T.; Bohlmann, J. The Molecular Basis for Wine Grape Quality—A Volatile Subject. Science 2006, 311, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Hardie, W.J.; Aggenbach, S.J.; Jaudzems, V.G. The Plastids of the Grape Pericarp and Their Significance in Isoprenoid Synthesis. Aust. J. Grape Wine Res. 1996, 2, 144–154. [Google Scholar] [CrossRef]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent Advances in the Transcriptional Regulation of the Flavonoid Biosynthetic Pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef]
- Ali, M.B.; Howard, S.; Chen, S.; Wang, Y.; Yu, O.; Kovacs, L.G.; Qiu, W. Berry Skin Development in Norton Grape: Distinct Patterns of Transcriptional Regulation and Flavonoid Biosynthesis. BMC Plant Biol. 2011, 11, 7. [Google Scholar] [CrossRef]
- Muganu, M.; Bellincontro, A.; Barnaba, F.E.; Paolocci, M.; Bignami, C.; Gambellini, G.; Mencarelli, F. Influence of Bunch Position in the Canopy on Berry Epicuticular Wax during Ripening and on Weight Loss during Postharvest Dehydration. Am. J. Enol. Vitic. 2011, 62, 91–98. [Google Scholar] [CrossRef]
- Chang, B.-M.; Keller, M. Cuticle and Skin Cell Walls Have Common and Unique Roles in Grape Berry Splitting. Hortic. Res. 2021, 8, 168. [Google Scholar] [CrossRef]
- Wada, H.; Matthews, M.A.; Shackel, K.A. Seasonal Pattern of Apoplastic Solute Accumulation and Loss of Cell Turgor during Ripening of Vitis Vinifera Fruit under Field Conditions. J. Exp. Bot. 2009, 60, 1773–1781. [Google Scholar] [CrossRef]
- Canaguier, A.; Grimplet, J.; Di Gaspero, G.; Scalabrin, S.; Duchêne, E.; Choisne, N.; Mohellibi, N.; Guichard, C.; Rombauts, S.; Le Clainche, I.; et al. A New Version of the Grapevine Reference Genome Assembly (12X.v2) and of Its Annotation (VCost.V3). Genom. Data 2017, 14, 56–62. [Google Scholar] [CrossRef]
- Brighenti, E.; Souza, A.L.K.; Brighenti, A.F.; Stefanini, M.; Trapp, O.; Gardin, J.P.P.; Caliari, V.; Dalbó, M.A.; Welter, L.J. Field Performance of Five White Pilzwiderstandsfahige (PIWI) Cultivars in the South of Brazil. Acta Hortic. 2019, 115–122. [Google Scholar] [CrossRef]
- Bavaresco, L.; Vercesi, A.; Belvini, P.; Dalla Costa, L.; Fogal, J.; Marcon, L.; Masaro, L.; Pascarella, G.; Pastro, M.; Pizzolato, A.; et al. Agronomic Performance of 21 New Disease Resistant Winegrape Varieties Grown in Northeast Italy. VITIS J. Grapevine Res. 2023, 81–87. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Jin, X.; Liu, L.; Dai, X.; Liu, Y.; Zhao, L.; Zheng, P.; Wang, X.; Liu, Y.; et al. Floral Transcriptomes Reveal Gene Networks in Pineapple Floral Growth and Fruit Development. Commun. Biol. 2020, 3, 500. [Google Scholar] [CrossRef] [PubMed]
- Shahan, R.; Zawora, C.; Wight, H.; Sittmann, J.; Wang, W.; Mount, S.M.; Liu, Z. Consensus Coexpression Network Analysis Identifies Key Regulators of Flower and Fruit Development in Wild Strawberry. Plant Physiol. 2018, 178, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, L.; Ming, M.; Hu, H.; Zhang, M.; Fan, J.; Song, B.; Zhang, S.; Wu, J. Comparative Transcriptomic Analysis Provides Insight into the Domestication and Improvement of Pear (P. pyrifolia) Fruit. Plant Physiol. 2019, 180, 435–452. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-L.; Wang, Z.-G.; Pei, M.-S.; Guo, L.-L.; Yu, Y.-H. Transcriptome Analysis Reveals Mechanism of Early Ripening in Kyoho Grape with Hydrogen Peroxide Treatment. BMC Genom. 2020, 21, 784. [Google Scholar] [CrossRef]
- Vannozzi, A.; Palumbo, F.; Magon, G.; Lucchin, M.; Barcaccia, G. The Grapevine (Vitis vinifera L.) Floral Transcriptome in Pinot Noir Variety: Identification of Tissue-Related Gene Networks and Whorl-Specific Markers in Pre- and Post-Anthesis Phases. Hortic. Res. 2021, 8, 200. [Google Scholar] [CrossRef]
- Zheng, H.; Dong, Y.; Nong, H.; Huang, L.; Liu, J.; Yu, X.; Zhang, Y.; Yang, L.; Hong, B.; Wang, W.; et al. VvSUN May Act in the Auxin Pathway to Regulate Fruit Shape in Grape. Hortic. Res. 2022, 9, uhac200. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, F.; Liu, G.; Abudureheman, R.; Bai, S.; Wu, X.; Zhang, C.; Ma, Y.; Wang, X.; Zha, Q.; et al. Transcriptome and Coexpression Network Analysis Reveals Properties and Candidate Genes Associated with Grape (Vitis vinifera L.) Heat Tolerance. Front. Plant Sci. 2023, 14, 1270933. [Google Scholar] [CrossRef]
- Botía, J.A.; Vandrovcova, J.; Forabosco, P.; Guelfi, S.; D’Sa, K.; Hardy, J.; Lewis, C.M.; Ryten, M.; Weale, M.E. The United Kingdom Brain Expression Consortium An Additional K-Means Clustering Step Improves the Biological Features of WGCNA Gene Co-Expression Networks. BMC Syst. Biol. 2017, 11, 47. [Google Scholar] [CrossRef]
- Wang, P.; Siao, W.; Zhao, X.; Arora, D.; Wang, R.; Eeckhout, D.; Van Leene, J.; Kumar, R.; Houbaert, A.; De Winne, N.; et al. Adaptor Protein Complex Interaction Map in Arabidopsis Identifies P34 as a Common Stability Regulator. Nat. Plants 2023, 9, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Heard, W.; Mbengue, M.; Robatzek, S. The INs and OUTs of Pattern Recognition Receptors at the Cell Surface. Curr. Opin. Plant Biol. 2012, 15, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Li, R.; Pan, J.; Ding, Z.; Lin, J. Endocytosis and Its Regulation in Plants. Trends Plant Sci. 2015, 20, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Garcia, E.; Tamang, T.M.; Park, J.; Dalby, M.; Martin-Urdiroz, M.; Rodriguez Herrero, C.; Vu, A.H.; Park, S.; Talbot, N.J.; Valent, B. Clathrin-Mediated Endocytosis Facilitates the Internalization of Magnaporthe Oryzae Effectors into Rice Cells. Plant Cell 2023, 35, 2527–2551. [Google Scholar] [CrossRef] [PubMed]
- Mbengue, M.; Bourdais, G.; Gervasi, F.; Beck, M.; Zhou, J.; Spallek, T.; Bartels, S.; Boller, T.; Ueda, T.; Kuhn, H.; et al. Clathrin-Dependent Endocytosis Is Required for Immunity Mediated by Pattern Recognition Receptor Kinases. Proc. Natl. Acad. Sci. USA 2016, 113, 11034–11039. [Google Scholar] [CrossRef]
- Spallek, T.; Beck, M.; Khaled, S.B.; Salomon, S.; Bourdais, G.; Schellmann, S.; Robatzek, S. ESCRT-I Mediates FLS2 Endosomal Sorting and Plant Immunity. PLOS Genet. 2013, 9, e1004035. [Google Scholar] [CrossRef]
- Postma, J.; Liebrand, T.W.H.; Bi, G.; Evrard, A.; Bye, R.R.; Mbengue, M.; Kuhn, H.; Joosten, M.H.A.J.; Robatzek, S. Avr4 Promotes Cf-4 Receptor-like Protein Association with the BAK1/SERK3 Receptor-like Kinase to Initiate Receptor Endocytosis and Plant Immunity. New Phytol. 2016, 210, 627–642. [Google Scholar] [CrossRef]
- Chung, K.P.; Zeng, Y.; Jiang, L. COPII Paralogs in Plants: Functional Redundancy or Diversity? Trends Plant Sci. 2016, 21, 758–769. [Google Scholar] [CrossRef]
- Kwon, C.; Bednarek, P.; Schulze-Lefert, P. Secretory Pathways in Plant Immune Responses. Plant Physiol. 2008, 147, 1575–1583. [Google Scholar] [CrossRef]
- Kulich, I.; Žárský, V. Autophagy-Related Direct Membrane Import from ER/Cytoplasm into the Vacuole or Apoplast: A Hidden Gateway Also for Secondary Metabolites and Phytohormones? IJMS 2014, 15, 7462–7474. [Google Scholar] [CrossRef]
- Ruano, G.; Scheuring, D. Plant Cells under Attack: Unconventional Endomembrane Trafficking during Plant Defense. Plants 2020, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Celebi, A.; Colak, N.; Hayirlioglu-Ayaz, S.; Kostadinović Veličkovska, S.; Ilieva, F.; Esatbeyoglu, T.; Ayaz, F.A. Accumulation of Phenolic Compounds and Antioxidant Capacity during Berry Development in Black ‘Isabel’ Grape (Vitis vinifera L. x Vitis labrusca L.). Molecules 2020, 25, 3845. [Google Scholar] [CrossRef] [PubMed]
- Phua, S.Y.; De Smet, B.; Remacle, C.; Chan, K.X.; Van Breusegem, F. Reactive Oxygen Species and Organellar Signaling. J. Exp. Bot. 2021, 72, 5807–5824. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, C.; Hao, N.; Fujiwara, T.; Wu, T. Endoplasmic Reticulum Stress and Reactive Oxygen Species in Plants. Antioxidants 2022, 11, 1240. [Google Scholar] [CrossRef] [PubMed]
- Kısaca, G.; Gazioglu Sensoy, R.I. Phenolic Contents, Organicacids and Antioxidant Capacities of Twenty Grape (Vitis vinifera L.) Cultivars Having Different Berry Colors. Food Meas. 2023, 17, 1354–1370. [Google Scholar] [CrossRef]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A Benchmark of Gene Expression Tissue-Specificity Metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- Ji, Z.; Wang, M.; Zhang, S.; Du, Y.; Cong, J.; Yan, H.; Guo, H.; Xu, B.; Zhou, Z. GDSL Esterase/Lipase GELP1 Involved in the Defense of Apple Leaves against Colletotrichum Gloeosporioides Infection. Int. J. Mol. Sci. 2023, 24, 10343. [Google Scholar] [CrossRef]
- Ding, L.-N.; Li, M.; Guo, X.-J.; Tang, M.-Q.; Cao, J.; Wang, Z.; Liu, R.; Zhu, K.-M.; Guo, L.; Liu, S.-Y.; et al. Arabidopsis GDSL1 Overexpression Enhances Rapeseed Sclerotinia Sclerotiorum Resistance and the Functional Identification of Its Homolog in Brassica Napus. Plant Biotechnol. J. 2020, 18, 1255–1270. [Google Scholar] [CrossRef]
- Wei, F.; Wing, R.A.; Wise, R.P. Genome Dynamics and Evolution of the Mla (Powdery Mildew) Resistance Locus in Barley. Plant Cell 2002, 14, 1903–1917. [Google Scholar] [CrossRef]
- Joshi, S.G.; Schaart, J.G.; Groenwold, R.; Jacobsen, E.; Schouten, H.J.; Krens, F.A. Functional Analysis and Expression Profiling of HcrVf1 and HcrVf2 for Development of Scab Resistant Cisgenic and Intragenic Apples. Plant Mol. Biol. 2011, 75, 579–591. [Google Scholar] [CrossRef]
- Rowland, O.; Ludwig, A.A.; Merrick, C.J.; Baillieul, F.; Tracy, F.E.; Durrant, W.E.; Fritz-Laylin, L.; Nekrasov, V.; Sjölander, K.; Yoshioka, H.; et al. Functional Analysis of Avr9/Cf-9 Rapidly Elicited Genes Identifies a Protein Kinase, ACIK1, That Is Essential for Full Cf-9–Dependent Disease Resistance in Tomato. Plant Cell 2005, 17, 295–310. [Google Scholar] [CrossRef]
- Toth, Z.; Winterhagen, P.; Kalapos, B.; Su, Y.; Kovacs, L.; Kiss, E. Expression of a Grapevine NAC Transcription Factor Gene Is Induced in Response to Powdery Mildew Colonization in Salicylic Acid-Independent Manner. Sci. Rep. 2016, 6, 30825. [Google Scholar] [CrossRef]
- Wang, P.; Liu, C.; Wang, D.; Liang, C.; Zhao, K.; Fan, J. Isolation of Resistance Gene Analogs from Grapevine Resistant to Downy Mildew. Sci. Hortic. 2013, 150, 326–333. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast One-Pass FASTQ Data Preprocessing, Quality Control, and Deduplication Using Fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. Gprofiler2—An R Package for Gene List Functional Enrichment Analysis and Namespace Conversion Toolset g:Profiler. F1000Research 2020, 9, ELIXIR-709. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID V3 | Gene ID V2 | Tau | Score | Mean Exp | Functional Annotation |
|---|---|---|---|---|---|
| Cabernet sauvignon | |||||
| Vitvi01g02070 | VIT_01s0127g00910 | 0.95 | 1.265 | 56.55 | AERO1 |
| Vitvi10g00903 | - | 1.00 | 1.249 | 23.71 | Leucin-rich repeat protein kinase |
| Vitvi01g02281 | VIT_01s0010g04010 | 1.00 | 1.182 | 9.77 | Unknown protein |
| Vitvi10g02153 | - | 1.00 | 1.133 | 5.16 | - |
| Vitvi07g00496 | VIT_07s0005g02310 | 0.93 | 1.133 | 12.47 | EXPA17 |
| Vitvi05g02072 | - | 0.93 | 1.130 | 11.93 | - |
| Vitvi10g00183 | - | 1.00 | 1.125 | 4.63 | - |
| Vitvi10g02415 | VIT_00s2472g00010 | 0.85 | 1.112 | 27.96 | Enhancer of mR-decapping protein 4 |
| Vitvi01g02068 | - | 1.00 | 1.111 | 3.84 | - |
| Vitvi10g02416 | - | 1.00 | 1.098 | 3.25 | - |
| Cabernet cortis | |||||
| Vitvi07g02026 | VIT_07s0130g00200 | 1.00 | 1.423 | 238.36 | VvGELP21-Lipase GDSL |
| Vitvi19g00082 | VIT_19s0014g01060 | 1.00 | 1.417 | 220.86 | Sesquiterpene synthase |
| Vitvi09g01530 | VIT_09s0002g01980 | 0.96 | 1.325 | 113.34 | Myosin-like protein XIK |
| Vitvi11g01266 | VIT_11s0052g01230 | 0.93 | 1.322 | 153.64 | Xyloglucan endotransglucosylase/hydrolase 23 |
| Vitvi09g01648 | - | 0.96 | 1.309 | 91.87 | - |
| Vitvi08g02288 | VIT_08s0007g04580 | 0.86 | 1.303 | 319.66 | UGT73C2 (UDP-glucosyl transferase 73C2) |
| Vitvi19g00324 | VIT_19s0014g04000 | 1.00 | 1.285 | 40.05 | Curculin (mannose-binding) lectin |
| Vitvi12g02451 | VIT_12s0134g00650 | 1.00 | 1.274 | 34.63 | Anthocyanin 5-aromatic acyltransferase |
| Vitvi19g01982 | VIT_19s0014g05140 | 0.95 | 1.271 | 64.65 | - |
| Vitvi15g00285 | VIT_15s0045g00270 | 1.00 | 1.265 | 30.67 | Serine/threonine-protein phosphatase BSL3 |
| Cabernet volos | |||||
| Vitvi14g00668 | VIT_14s0036g00990 | 0.86 | 1.432 | 1814.13 | Polyubiquitin (UBQ4) |
| Vitvi11g01637 | VIT_11s0052g00270 | 0.85 | 1.346 | 652.97 | R protein MLA10 |
| Vitvi11g00879 | VIT_11s0065g00040 | 0.89 | 1.331 | 321.15 | CYP706A12 |
| Vitvi04g00345 | VIT_04s0008g04000 | 0.87 | 1.295 | 256.39 | Unknown |
| Vitvi03g01478 | VIT_03s0038g04230 | 0.88 | 1.240 | 103.63 | Dihydroflavonol 4-reductase |
| Vitvi08g02374 | VIT_08s0007g07760 | 0.93 | 1.175 | 21.69 | Polygalacturonase PG1 |
| Vitvi16g01677 | - | 1.00 | 1.169 | 8.42 | - |
| Vitvi08g00789 | VIT_08s0058g00650 | 1.00 | 1.161 | 7.72 | Aldose reductase |
| Vitvi11g01568 | VIT_11s0065g00740 | 1.00 | 1.142 | 6.04 | A -phase-promoting complex subunit 8 |
| Vitvi01g01642 | VIT_01s0010g03550 | 1.00 | 1.140 | 5.97 | Nuclear transcription factor Y sub-B related |
| Sauvignon blanc | |||||
| Vitvi04g00029 | VIT_04s0008g00370 | 0.85 | 1.156 | 57.39 | Clavata1 receptor kinase (CLV1) |
| Vitvi06g01648 | VIT_06s0004g02550 | 0.85 | 1.150 | 52.71 | Kiwellin Ripening-related protein grip22 |
| Vitvi04g00021 | VIT_04s0008g00300 | 0.85 | 1.147 | 50.92 | Clavata1 receptor kinase (CLV1) |
| Vitvi09g01948 | - | 0.85 | 1.140 | 45.92 | HcrVf2 protein |
| Vitvi03g00460 | VIT_03s0063g01000 | 0.85 | 1.127 | 39.01 | Blue (type 1) copper domain |
| Vitvi07g01769 | VIT_07s0031g00850 | 0.87 | 1.094 | 20.80 | Patatin |
| Vitvi01g01852 | VIT_01s0011g00990 | 0.87 | 1.080 | 17.09 | RPM1 |
| Vitvi10g00005 | VIT_10s0116g00150 | 0.87 | 1.078 | 16.85 | Receptor kinase RK20-1 |
| Vitvi00g02077 | VIT_00s0895g00010 | 0.87 | 1.061 | 13.52 | Glucan 1,3-beta-glucosidase |
| Vitvi16g02124 | VIT_00s0294g00100 | 1.00 | 1.060 | 2.29 | BR insensitive 1 receptor kinase 1 |
| Sauvignon nepis | |||||
| Vitvi09g01181 | VIT_09s0018g00780 | 1.00 | 1.506 | 710.79 | HcrVf1 protein |
| Vitvi18g02399 | VIT_18s0089g01040 | 0.97 | 1.474 | 647.26 | Avr9 elicitor response |
| Vitvi10g01863 | VIT_10s0003g03530 | 0.97 | 1.427 | 382.88 | Lupeol synthase |
| Vitvi10g01875 | VIT_10s0003g03650 | 0.97 | 1.427 | 381.69 | Beta-amyrin synthase |
| Vitvi13g02352 | VIT_13s0139g00190 | 0.92 | 1.408 | 529.73 | Disease resistance protein RGA2 |
| Vitvi12g02393 | VIT_12s0059g01790 | 0.91 | 1.356 | 321.51 | Caffeic acid O-methyltransferase |
| Vitvi02g00721 | VIT_02s0012g01610 | 0.87 | 1.355 | 506.49 | Beta-1,3-gluca -se precursor |
| Vitvi03g01757 | - | 1.00 | 1.314 | 63.03 | - |
| Vitvi03g00910 | VIT_03s0167g00050 | 0.92 | 1.298 | 140.99 | Conca-valin A lectin |
| Vitvi16g00665 | VIT_16s0022g00420 | 0.96 | 1.294 | 81.21 | SRG1 oxidoreductase |
| Sauvignon rytos | |||||
| Vitvi01g01410 | - | 1.00 | 1.326 | 67.07 | - |
| Vitvi08g00957 | VIT_08s0040g00920 | 0.87 | 1.280 | 207.61 | Glutathione S-transferase 25 GSTU7 |
| Vitvi14g00080 | VIT_14s0060g00990 | 1.00 | 1.269 | 31.94 | Unknown |
| Vitvi13g02566 | VIT_13s0156g00390 | 1.00 | 1.267 | 31.10 | Myb family |
| Vitvi18g03265 | VIT_18s0089g01000 | 0.95 | 1.256 | 51.80 | F-box family protein |
| Vitvi15g01230 | - | 1.00 | 1.229 | 19.32 | - |
| Vitvi15g01425 | VIT_15s0021g01450 | 1.00 | 1.224 | 18.06 | No hit |
| Vitvi13g01636 | VIT_13s0158g00050 | 1.00 | 1.223 | 17.85 | Serine carboxypeptidase |
| Vitvi10g01830 | VIT_10s0003g02420 | 1.00 | 1.217 | 16.27 | SRG1oxidoreductase |
| Vitvi17g00462 | - | 1.00 | 1.216 | 16.08 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scariolo, F.; Gabelli, G.; Magon, G.; Palumbo, F.; Pirrello, C.; Farinati, S.; Curioni, A.; Devillars, A.; Lucchin, M.; Barcaccia, G.; et al. The Transcriptional Landscape of Berry Skin in Red and White PIWI (“Pilzwiderstandsfähig”) Grapevines Possessing QTLs for Partial Resistance to Downy and Powdery Mildews. Plants 2024, 13, 2574. https://doi.org/10.3390/plants13182574
Scariolo F, Gabelli G, Magon G, Palumbo F, Pirrello C, Farinati S, Curioni A, Devillars A, Lucchin M, Barcaccia G, et al. The Transcriptional Landscape of Berry Skin in Red and White PIWI (“Pilzwiderstandsfähig”) Grapevines Possessing QTLs for Partial Resistance to Downy and Powdery Mildews. Plants. 2024; 13(18):2574. https://doi.org/10.3390/plants13182574
Chicago/Turabian StyleScariolo, Francesco, Giovanni Gabelli, Gabriele Magon, Fabio Palumbo, Carlotta Pirrello, Silvia Farinati, Andrea Curioni, Aurélien Devillars, Margherita Lucchin, Gianni Barcaccia, and et al. 2024. "The Transcriptional Landscape of Berry Skin in Red and White PIWI (“Pilzwiderstandsfähig”) Grapevines Possessing QTLs for Partial Resistance to Downy and Powdery Mildews" Plants 13, no. 18: 2574. https://doi.org/10.3390/plants13182574
APA StyleScariolo, F., Gabelli, G., Magon, G., Palumbo, F., Pirrello, C., Farinati, S., Curioni, A., Devillars, A., Lucchin, M., Barcaccia, G., & Vannozzi, A. (2024). The Transcriptional Landscape of Berry Skin in Red and White PIWI (“Pilzwiderstandsfähig”) Grapevines Possessing QTLs for Partial Resistance to Downy and Powdery Mildews. Plants, 13(18), 2574. https://doi.org/10.3390/plants13182574