



Cells 2024, 13(17), 1479; https://doi.org/10.3390/cells13171479 - 3 Sep 2024
Viewed by 1106
Abstract
►
Show Figures
LMNA-related dilated cardiomyopathy (DCM) is an autosomal-dominant genetic condition with cardiomyocyte and conduction system dysfunction often resulting in heart failure or sudden death. The condition is caused by mutation in the Lamin A/C (LMNA) gene encoding Type-A nuclear lamin proteins
[...] Read more.
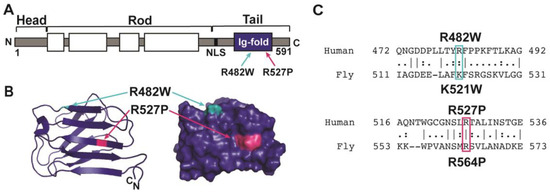
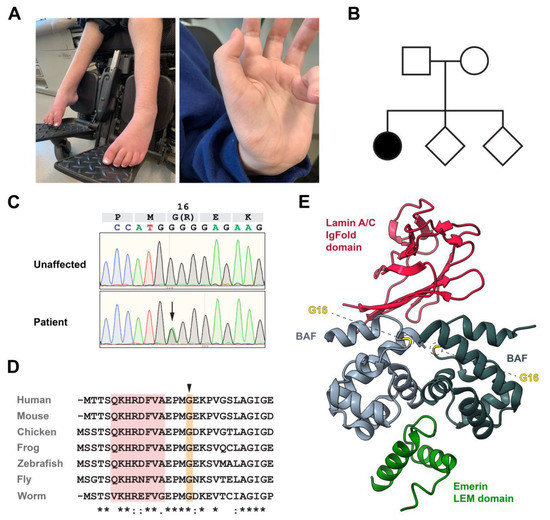
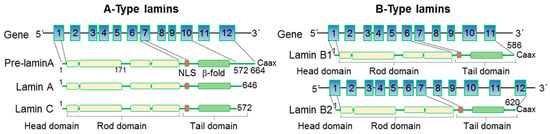
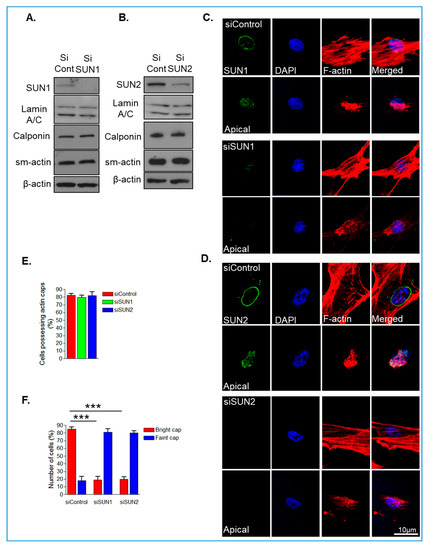
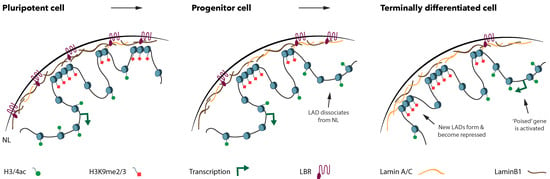
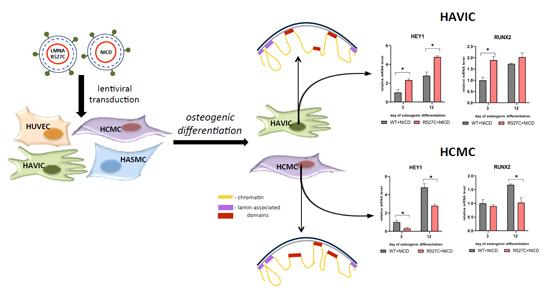
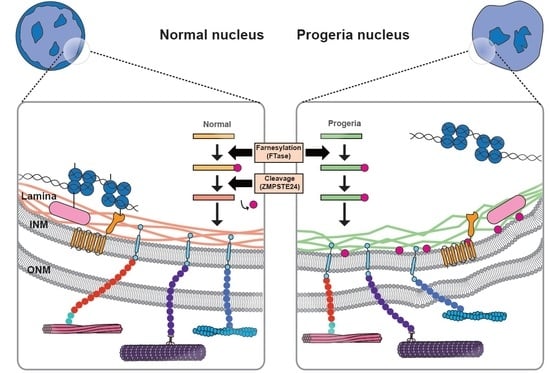
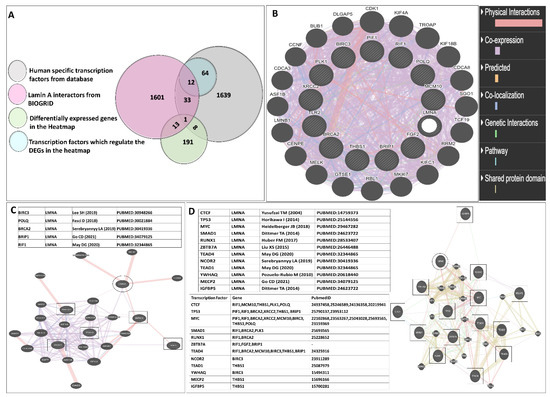
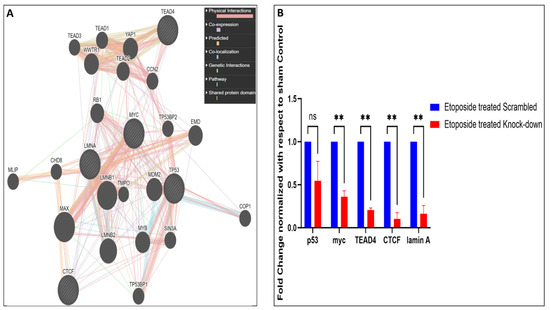
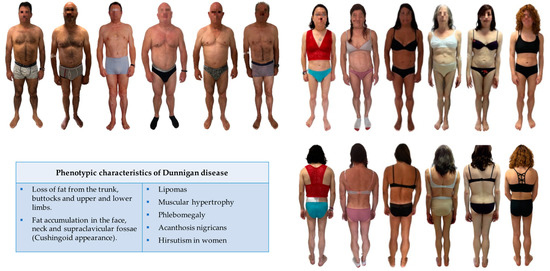
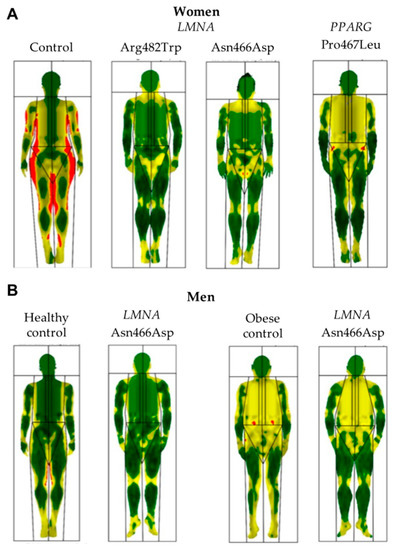
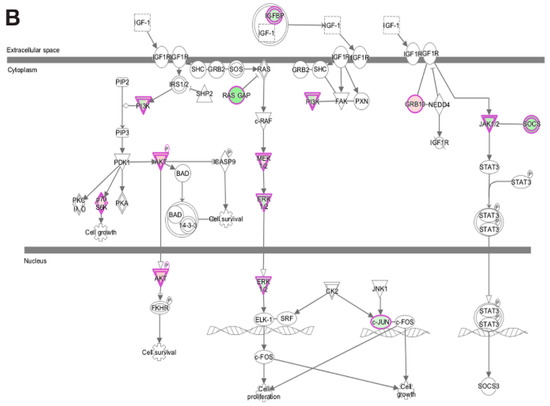
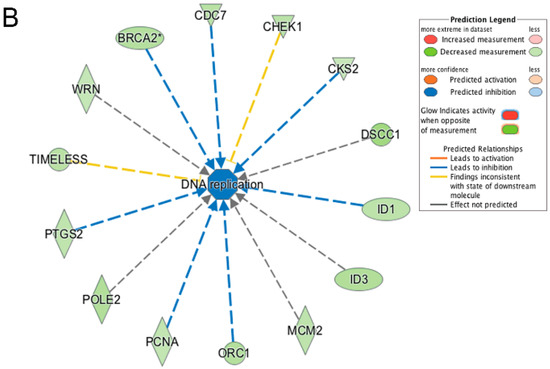
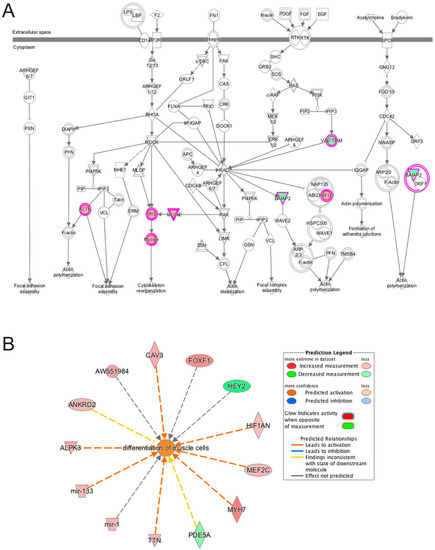
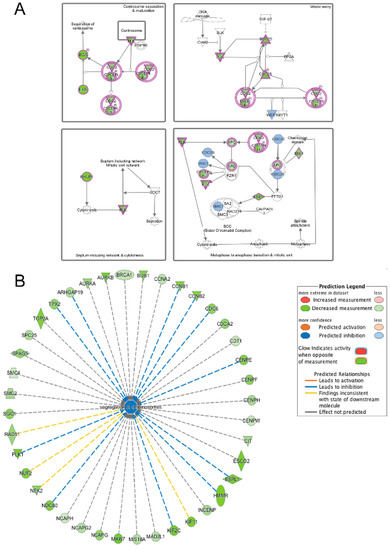
LMNA-related dilated cardiomyopathy (DCM) is an autosomal-dominant genetic condition with cardiomyocyte and conduction system dysfunction often resulting in heart failure or sudden death. The condition is caused by mutation in the Lamin A/C (LMNA) gene encoding Type-A nuclear lamin proteins involved in nuclear integrity, epigenetic regulation of gene expression, and differentiation. The molecular mechanisms of the disease are not completely understood, and there are no definitive treatments to reverse progression or prevent mortality. We investigated possible mechanisms of LMNA-related DCM using induced pluripotent stem cells derived from a family with a heterozygous LMNA c.357-2A>G splice-site mutation. We differentiated one LMNA-mutant iPSC line derived from an affected female (Patient) and two non-mutant iPSC lines derived from her unaffected sister (Control) and conducted single-cell RNA sequencing for 12 samples (four from Patients and eight from Controls) across seven time points: Day 0, 2, 4, 9, 16, 19, and 30. Our bioinformatics workflow identified 125,554 cells in raw data and 110,521 (88%) high-quality cells in sequentially processed data. Unsupervised clustering, cell annotation, and trajectory inference found complex heterogeneity: ten main cell types; many possible subtypes; and lineage bifurcation for cardiac progenitors to cardiomyocytes (CMs) and epicardium-derived cells (EPDCs). Data integration and comparative analyses of Patient and Control cells found cell type and lineage-specific differentially expressed genes (DEGs) with enrichment, supporting pathway dysregulation. Top DEGs and enriched pathways included 10 ZNF genes and RNA polymerase II transcription in pluripotent cells (PP); BMP4 and TGF Beta/BMP signaling, sarcomere gene subsets and cardiogenesis, CDH2 and EMT in CMs; LMNA and epigenetic regulation, as well as DDIT4 and mTORC1 signaling in EPDCs. Top DEGs also included XIST and other X-linked genes, six imprinted genes (SNRPN, PWAR6, NDN, PEG10, MEG3, MEG8), and enriched gene sets related to metabolism, proliferation, and homeostasis. We confirmed Lamin A/C haploinsufficiency by allelic expression and Western blot. Our complex Patient-derived iPSC model for Lamin A/C haploinsufficiency in PP, CM, and EPDC provided support for dysregulation of genes and pathways, many previously associated with Lamin A/C defects, such as epigenetic gene expression, signaling, and differentiation. Our findings support disruption of epigenomic developmental programs, as proposed in other LMNA disease models. We recognized other factors influencing epigenetics and differentiation; thus, our approach needs improvement to further investigate this mechanism in an iPSC-derived model.
Full article

Graphical abstract










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}